
New insights into the qualitative phenolic profile of Ficus carica L. fruits and leaves from Tunisia using ultra-high-performance liquid chromatography coupled to quadrupole-time-of-flight mass spectrometry and their antioxidant activity†
Abstract
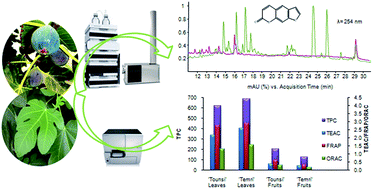
Ficus carica L. fruits have been consumed from the earliest times, and other parts of the tree have been used for traditional medicinal purposes. Nowadays, the beneficial properties of this and other Ficus species are attributed to the presence of key phytochemicals. To increase our knowledge about this topic, the present study has conducted phenolic profiling of the leaves and whole fruits from two Tunisian cultivars, ‘Temri’ and ‘Tounsi’, using reversed-phase ultra-high-performance liquid chromatography (RP-UHPLC) coupled to two detection systems: diode-array detection (DAD) and quadrupole time-of-flight (QTOF) mass spectrometry (MS). UV-Vis absorption was a valuable tool for classifying phenolic compounds into families, while MS using electrospray ionization (ESI) and MS/MS allowed the molecular formula to be established and structural information to be obtained. The total phenol content and the antioxidant activity were also assessed. As a result, in the negative ionization mode 91 phenolic compounds were characterized including hydroxybenzoic acids, hydroxycinnamic acids, hydroxycoumarins and flavanoids (flavonols, flavones, flavanones, flavanonols, flavanols and isoflavones). This work was complemented by the detection of other 18 phenolic compounds in the positive ionization mode, including anthocyanins and furanocoumarins. To the best of our knowledge, this is the first time most of these compounds have been tentatively reported in F. carica. These results indicate the complexity of this family of secondary metabolites in F. carica, as well as the potential of this analytical method for characterization purposes. In conclusion, the qualitative phenolic profile, total phenolic content and antioxidant activity differed especially between leaves and fruits.
 Please wait while we load your content...
Please wait while we load your content...

