
Ultrasonic-assisted self-assembly synthesis of highly dispersed β-MnO2 nano-branches interwoven with graphene flakes for catalytic oxidation of aromatic compounds†
Jun Mei and
Long Zhang*
Jilin Provincial Engineering Laboratory for the Complex Utilization of Petro-Resources and Biomass, Changchun University of Technology, Changchun 130012, P R China. E-mail: zhanglongzhl@163.com; Fax: +86-431-8571-6328; Tel: +86-431-8571-7216
First published on 26th January 2015
Abstract
A controllable ultrasonic-assisted self-assembly process is used for the fabrication of novel β-MnO2 nano-branched–graphene composites by branched β-MnO2 interwoven with flake graphene. The composites demonstrate high activity in catalytic oxidation of a methylene blue (MB) model. Roles of different components in the composites were confirmed and the optimum reaction conditions were also investigated. By cyclic tests, the composites show excellent chemical stability. Finally, the catalytic oxidation steps were further studied by analysis of the products. These results will contribute to the wide applications of the new composites in the catalytic oxidation of other aromatic compounds.
1. Introduction
Aromatic compounds, the main components of dye-containing wastewater, have become serious pollutants with industrial developments, e.g., in textile, paper, plastic, leather, food and cosmetic industries. Most types of these compounds are of high chroma, toxicity and chemical stability.1–3 Treatment of these compounds is of special importance and more efficient technologies are urgently needed. Catalytic oxidation, based on chemical reactions to make aromatic compounds oxidize into smaller organic or inorganic molecules, emerged as a simple and effective strategy.4–11 In particular, the cost-effective advanced catalytic oxidation processes (AOPs) are suitable for different kinds of aromatic compounds without selectivity, which are dependent on the generation of reactive species such as hydroxyl radicals (˙OH).12–16 Unfortunately, the shortage of efficient catalysts has greatly limited their application scope.In this respect, compared with commercial micro-sized powders, MnO2 nanoparticles with good physical and chemical properties have become attractive candidates for heterogeneous catalysis. Different crystalline forms, appearances and sizes of MnO2 nanoparticles have been synthesized for catalytic oxidation of aromatic compounds, including α/β-MnO2 nanorods, nanowires and nanospheres.17–24 However, due to the nature of agglomeration, the catalytic activity and efficiency of MnO2 nanoparticles are still limited, usually accompanying with serious pulverization phenomena. With the reaction carries through, the activity is decreasing, leading to long reaction time or harsh reaction conditions needed. Therefore, improving dispersion of MnO2 nanoparticles is an urgent problem to be solved.
In recent years, research interest on graphene has been increasing in many fields.25–29 Unique flake-like structured graphene, especially chemically modified graphene, with high specific surface area and functional groups for chemical activity, has been an important and low-cost catalyst support for nanoparticles loading. It has been proven that the electrochemical performances of MnO2 nanoparticles can be enhanced by incorporation of highly conductive materials.30–33 Thus, the catalytic performances may also be improved by binding MnO2 nanoparticles with graphene flakes. There are also several reports on the synthesis of MnO2 nanoparticles and graphene composites by hydrothermal reducing method for catalytic oxidation.34,35 However, the existing results are not satisfying, with a longer reaction time (about 300 min) or a higher temperature (up to 50 °C) needed. The poor dispersion of MnO2 nanoparticles on the surface of graphene flakes has a great effect on their catalytic activity and cyclic stability. This was probably related to the assembly process of MnO2 nanoparticles and graphene. Besides, the agglomeration of graphene flakes is another great obstacle.
In our work, novel β-MnO2 nano-branched–graphene composites were synthesized by highly dispersed MnO2 nanoparticles interwoven with graphene flakes. In comparison with nanorods, nanowires and nanospheres, different dimensional MnO2 nano-branches are easier to be assembled with graphene flakes. Under the assistance of ultrasonic wave, the branched MnO2 nanoparticles, just like needle and thread, were interwoven with graphene flakes by a self-assembly process. This process is similar to that in knitting a pair of gloves for wearing. By this method, MnO2 nano-branches can be dispersed easily and the agglomeration of graphene flakes can also be avoided. The as-obtained composites are chemically and thermally stable. For catalytic oxidation activity tests, methylene blue (MB), a heterocyclic aromatic compound mainly used in a range of biology and chemistry fields, was chosen as a model. The reaction was conducted in neutral condition at room temperature with peroxide (H2O2) as oxidant. Here, a new adsorbent–catalyst system is constructed with graphene of high specific surface as the adsorbent and β-MnO2 nano-branches of high catalytic activity as the catalyst (Fig. 1). In terms of adsorption and catalytic efficiency, this catalytic system demonstrates excellent catalytic oxidation performances.
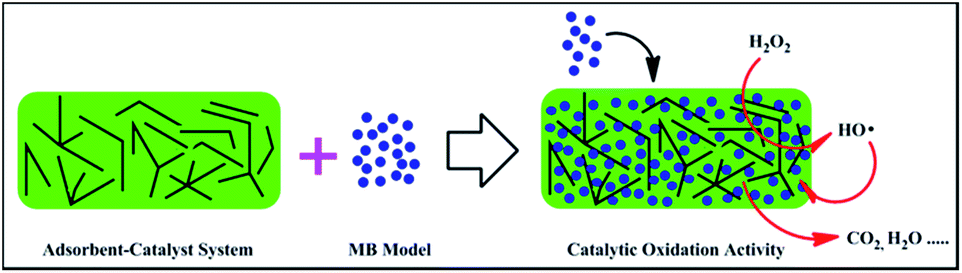 | ||
| Fig. 1 Schematic illustration of the new adsorbent–catalyst system in β-MnO2 nano-branched–graphene composites. | ||
2. Experimental section
2.1. Reagents and materials
Natural flake graphite (500 meshes) was purchased from Shanghai Yifan graphite Ltd., China. Potassium permanganate (KMnO4), concentrated sulfuric acid (H2SO4, 98%), peroxide (H2O2, 30 wt%), hydrochloric acid (HCl, 1.0 mol L−1), polyethylene glycol (PEG200, average molecular weight 200) and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd., China. All chemicals are analytical grade and used without further purification. Water used in the study was distilled for three times and purified by a Milli-Q system.2.2. Preparation of graphene oxide (GO) suspension
The graphite oxide was prepared according to a modified Hummers method.36,37 Briefly, natural flake graphite (2.0 g) were added into H2SO4 (90 mL) and stirred in an ice bath for 10 min. Then, KMnO4 (12.0 g) was slowly added and the mixture was maintained at 50 °C for 6 hours. Followed by adding 160 mL of water, the reaction was kept at 80 °C for 30 min. Then, 12 mL of H2O2 was added and the solid was washed by hydrochloric acid and water repeatedly. After dried at 60 °C overnight, the brown powder was re-dispersed in water by ultrasonic method at a power of 280 W for 90 min to form a homogeneous brown suspension with a concentration of about 1.0 mg mL−1.2.3. Preparation of MnOOH precursor
To synthesize MnOOH precursor, KMnO4 (50.0 mg) was dissolved into water (25 mL) at room temperature. After 5 min, PEG200 (2 mL) was added and the mixture was stirred for 30 min. The obtained solution was transferred into a Teflon-lined stainless steel autoclave (30 mL) and was kept to be heated to 160 °C for 12 h. After cooling to room temperature, the formed precipitate was filtered and washed with water and ethanol for three times, respectively. The product was vacuum-dried for 12 hours.2.4. Preparation of β-MnO2 nano-branches
The as-produced MnOOH precursor was placed in a crucible and heated in a furnace at 350 °C for 4 hours to form black β-MnO2 nano-branches.2.5. Synthesis of β-MnO2 nano-branched–graphene composites
The composites with different mass fractions of β-MnO2 nano-branches were synthesized. Various amounts of MnO2 were added into the above GO suspension under ultrasonic vibration at a power of 350 W for 60 min. Then, the as-made mixture was transferred into a Teflon-lined stainless steel autoclave and kept heated at 100 °C for 10 h. After cooling to room temperature, black products were obtained by filtration, washing and freeze-drying. The as-synthesized composites were denoted as MnO2/GO-X, where X represents the mass fraction of MnO2 in the feeding process. In our study, MnO2/GO-33, MnO2/GO-75 and MnO2/GO-94 were synthesized, and the feeding mass ratios of MnO2/GO are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 3:1 and 15:1, respectively.
:2, 3:1 and 15:1, respectively.
2.6. Characterization
The as-prepared samples were tested in a Rigaku D/max-2500 X-ray powder diffractometer using Cu Kα radiation (λ = 0.1542 nm) with scattering angles (2θ) of 5–80°, operating at 40 kV and a cathode current of 30 mA. Thermogravimetric analysis (TGA) was carried out in a SDT-Q600 analyzer under air atmosphere at a temperature range of 25–800 °C with a heating rate of 5 °C min−1. Morphologies of the samples were characterized by scanning electron microscopy (SEM, JEOL, JSM-5500LV, 30 kV, 10−5 Torr) and transmission electron microscopy (TEM, JEOL, JEM-2000EX, 200 kV, 10−7 Torr). All samples for TEM tests were prepared by dropping a suspension of each sample (dispersed in ethanol earlier using an ultrasonic bath for 15 min) onto the carbon Holey grid (JEOL, 400 meshes). Specific surface area measurements for all samples were performed by nitrogen adsorption at 77 K with a Micrometric ASAP 2020 physisorption analyzer. Samples were outgassed for 6 h under vacuum at 350 °C before adsorption. X-ray photoelectron spectroscopy (XPS) was done by a Thermo Escalab 250 with a monochromatic AlKα source at room temperature with the samples cleaned before. All the binding energies were referenced to the C 1s peak at 284.8 eV.2.7. Catalytic oxidation activity tests
In a typical test of MB model, H2O2 solution (20 mL) was added into MB dye solution (1000 mL, 25 mg L−1) at room temperature. Various amounts of the as-made catalysts were then added to the mixture under continuous stirring. To avoid photo-degradation, the reactions were carried out in a brown flask wrapped with a tin foil paper. At different time intervals, 1 mL aliquots of the suspension were pipetted out, filtered through 0.22 μm membrane, and diluted into a volumetric flask (25 mL). After reaction, the suspension was centrifuged at 10000 rpm for 10 min and the residual solid was separated and dried at 60 °C for 12 hours. Each activity test was repeated three times at the same reaction conditions and the reported data are the average values of each three test cases.
The catalytic oxidation process of MB solution was monitored by UV-Vis spectroscopy (UV1901, Shanghai Youke Instrument Co., Ltd.). All samples taken at different time intervals were analyzed in an optical quartz cell (Path length 1.0 cm) with a reference aqueous solution. Concentrations were measured by the absorbance intensity at the maximum absorption wavelength (664 nm) according to the Beer's law. The discoloration efficiency of MB solution was calculated from the following equation:

3. Results and discussion
3.1. Self-assembly processes of β-MnO2 nano-branched–graphene composites
Ultrasonic-assisted self-assembly routines (Fig. 2) were used to synthesize the β-MnO2 nano-branched–graphene composites. Initially, branched MnOOH precursor was prepared with PEG200 as capping organic molecules, which is crucial for regulation of crystal nucleation and growth process in the hydrothermal reaction system. The production yield (97.2%) of the MnOOH precursor was shown to be high and the process was easy for scale-up. Then, β-MnO2 nano-branches were formed after calcination of MnOOH precursor at 350 °C in air ambient. Simultaneously, GO suspension was obtained by the modified Hummers method, which is easy to operate in the solution. The basal plane of obtained GO sheet was decorated by hydroxyl and epoxy functional groups and edges by carboxyl and carbonyl groups. And then, under the assistance of ultrasonic wave, β-MnO2 nano-branches were self-assembled with GO flakes successfully. Relying on the ultrasonic cavitation effects, β-MnO2 nano-branches were highly dispersed and interwoven with flake-like GO. During this process, strong chemical bonds, such as Mn–O–Mn, may be formed between β-MnO2 nano-branches and the oxygen-containing functional groups of GO flakes. Besides, hydroxyl groups adsorbed on the surface of β-MnO2 nano-branches can form hydrogen bond with hydroxyl, carboxyl and carbonyl groups on the surfaces of GO flakes. Moreover, the electrostatic force was also beneficial to enhance the dispersibility and stability. Depending on ultrasonic energy, a knitting skill was conducted between β-MnO2 nano-branches, as needle and thread, and GO flakes, as pieces of cloth. Finally, in order to improve electrical conductivity, thermal treatment is essential for the synthesis of β-MnO2 nano-branched–graphene composites, which contributes to electron mobility and facilitates electron transfer over catalytic reactions. Many oxygen-containing functional groups were removed, particularly from the basal plane. The as-synthesized composites with stable structure demonstrate high catalytic oxidation activity and cyclic stability. | ||
| Fig. 2 Schematic illustration of the preparation processes of β-MnO2 nano-branched–graphene composites. | ||
3.2. Structural characterization
It is confirmed by XRD analyses (Fig. 3a) that MnOOH, MnO2, MnO2/GO-33, MnO2/GO-75, MnO2/GO-94 composites are crystalline. The MnOOH precursor is pure and holds a monoclinic structure of MnOOH with SG P21/c (no. 14) and lattice parameters of a = 5.300 Å, b = 5.278 Å, c = 5.307 Å, β = 114.36° (JCPDS card no. 41-1379). Following calcination of MnOOH at 350 °C for 4 hours, the diffraction peaks at 2θ = 28.64°, 37.32°, 42.77°, 56.62° and 59.30° can be ascribed to the reflections of (1 1 0), (1 0 1), (1 1 1), (2 1 1) and (2 2 0) planes, which were indexed to β-MnO2, representing a tetragonal phase (SG P42/mnm, no. 136) with lattice constants of a = 4.399 Å, c = 2.874 Å (JCPDS card no. 24-0735). | ||
| Fig. 3 (a) XRD patterns and (b) TGA curves of the samples. | ||
The MnO2/GO-33, MnO2/GO-75 and MnO2/GO-94 composites show identical peaks with those of MnO2, which verifies that the MnO2 phase remains unchanged in the composites. Since MnO2 crystals are interwoven with a majority of graphene flakes, the MnO2/GO-33 composites showed poor crystallinity, just as enough pieces of cloth with a little needle and thread. A broad peak at around 24° is attributed to the agglomeration of graphene flakes, corresponding to d-spacing of 0.37 nm, similar to that of the pristine graphite (0.34 nm). By increasing the mass fraction of MnO2, the peak located at around 24° disappeared. This can prove that by a gradual increase in MnO2 nano-branches, agglomeration of graphene flakes has been prevented effectively. Considering that the content and aggregation form of GO can affect its diffractive peaks, there was no peak detected at around 10° (the (001) crystal plane of GO) for the composites.
The composition and thermal stability of the as-produced β-MnO2 nano-branched–graphene composites were tested by TGA (Fig. 3b). As reported in,35,38 weight loss below 300 °C is attributed to the removal of physical adsorbed water, water in the lattice and the residual oxygen-containing functional groups in the composites. As the temperature increased to above 380 °C, the graphene portion of the composites has been oxidized by the air flow and the carbon sketch has been removed completely. The remained mass at above 800 °C is attributed to the phase transformation of MnO2, which turns into Mn2O3. The mass ratios of MnO2 in the composites, listed in Table 1, are estimated by the remained mass ratios (Mn2O3) when the composites are oxidized in air ambient. The difference between the calculated mass ratios and feeding ratios is attributed to the removal of oxygen-containing functional groups.
| Samples | Feeding ratios (MnO2/GO) | Calculated mass ratios (%) | Specific surface area (m2 g−1) |
|---|---|---|---|
| MnOOH | — | — | 2.89 |
| MnO2 | — | — | 12.20 |
| MnO2/GO-33 | 1:2 |
38.5 | 87.43 |
| MnO2/GO-75 | 3:1 |
82.3 | 102.11 |
| MnO2/GO-94 | 15:1 |
96.5 | 93.58 |
XPS has been used to further elucidate the chemical composition of β-MnO2 nano-branched–graphene composites. The peaks associated with Mn, C and O can be observed in the survey spectrum (Fig. 4a). The Mn 2p3/2 and Mn 2p1/2 peaks are centered at 642.2 eV and 653.9 eV, respectively (Fig. 4b). This indicates the presence of MnO2 (Mn4+) in the composites with a spin energy separation of 11.7 eV, in good accordance with the previous data on MnO2.39 From the C 1s deconvolution spectrum of GO (Fig. S1, ESI†), three peaks centered at 284.6, 286.7, and 288.6 eV are shown, which are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C in aromatic rings, C–O and CO groups, respectively. The C–O group is classified as epoxy/alkoxy.40 However, for the composites (Fig. 4c), the peak intensity for CC/C–C (284.6 eV) is higher and the intensity of the peaks associated with oxygen-containing functional groups (C–O and CO) is lower. This shows that most of the oxygen-containing functional groups have been removed. The β-MnO2 nano-branched–graphene composites have been obtained with GO flakes reduced to graphene. Fig. 4d shows the O 1s spectrum, with peaks centered at 529.0 and 532.2 eV attributed to the Mn–O–Mn and C–O/CO groups, respectively. The Mn atoms may interact with O atoms of the residual oxygen-containing functional groups to generate a Mn–O–Mn group.35,41 The stable combination of β-MnO2 nano-branches and graphene, via physical adsorption or a chemical reaction (by a hydrogen bond or a covalent coordination bond), may ease the contingent change of MnO2 nano-branches to enhance the cycling performance during the catalytic reactions.
C/C–C in aromatic rings, C–O and CO groups, respectively. The C–O group is classified as epoxy/alkoxy.40 However, for the composites (Fig. 4c), the peak intensity for CC/C–C (284.6 eV) is higher and the intensity of the peaks associated with oxygen-containing functional groups (C–O and CO) is lower. This shows that most of the oxygen-containing functional groups have been removed. The β-MnO2 nano-branched–graphene composites have been obtained with GO flakes reduced to graphene. Fig. 4d shows the O 1s spectrum, with peaks centered at 529.0 and 532.2 eV attributed to the Mn–O–Mn and C–O/CO groups, respectively. The Mn atoms may interact with O atoms of the residual oxygen-containing functional groups to generate a Mn–O–Mn group.35,41 The stable combination of β-MnO2 nano-branches and graphene, via physical adsorption or a chemical reaction (by a hydrogen bond or a covalent coordination bond), may ease the contingent change of MnO2 nano-branches to enhance the cycling performance during the catalytic reactions.
 | ||
| Fig. 4 (a) XPS survey spectra; (b) Mn 2p; (c) C 1s and (d) O 1s spectra of the as-obtained β-MnO2 nano-branched–graphene composites. | ||
SEM and TEM studies have also been conducted to reveal the morphology of the samples in more details. The SEM image of GO is shown in Fig. 5a. It displays a smooth plane with strands of wrinkles, implying that graphite has been stripped into graphene flakes and also oxidized into GO. As shown in Fig. 5b and 6a, the branched MnOOH precursor presents different dimensional features with a length of 500 nm to several micrometers. After calcination of the precursor, the β-MnO2 nano-branched products (Fig. 5c, 6b and c) maintained their original morphology but obtained a higher specific surface area (12.20 m2 g−1, Table 1). The removal of hydrogen atoms affects the particles size and the branches of β-MnO2 become much slender. This specific branched structure makes it a good candidate for assembling with flake graphene, which is easy to stack into layered construction.
 | ||
| Fig. 5 SEM images of (a) GO, (b) MnOOH precursor, (c) β-MnO2 nano-branches, (d) MnO2/GO-33, (e) MnO2/GO-75 and (f) MnO2/GO-94 composites. Scale bars: 2 μm. | ||
 | ||
| Fig. 6 TEM images on (a) MnOOH precursor, (b and c) β-MnO2 nano-branches, (d) MnO2/GO-33, (e) MnO2/GO-75 and (f) MnO2/GO-94 composites. Scale bars: 500 nm. | ||
The SEM and TEM images of β-MnO2 nano-branched–graphene composites with different mass fractions of MnO2 (MnO2/GO-33, MnO2/GO-75 and MnO2/GO-94) are displayed in Fig. 5d–f and 6d–f. As the mass fractions of MnO2 increase, the morphology of the products changes dramatically. In the MnO2/GO-33 composites (Fig. 5d and 6d), a small amount of MnO2 nano-branches was interwoven with relatively large amounts of graphene flakes, making the graphene flakes easy for agglomeration, which is in accordance with XRD analysis. When the ratio is 75%, MnO2 nano-branches are well-distributed on the graphene flakes (Fig. 5e and 6e). In these composites, enough MnO2 nano-branches were provided as needle and thread for interweaving graphene flakes, and graphene flakes also change the distribution state of MnO2 nano-branches. This wonderful combinations result in the highest value of specific surface area (102.11 m2 g−1), higher than that of reported composites (49 m2 g−1).35 For ratios up to 94%, a little amount of graphene flakes was interwoven with MnO2 nano-branches (Fig. 5f and 6f). The agglomeration of MnO2 nano-branches becomes serious and the specific area is lower (93.58 m2 g−1), yet still much higher than that of pure MnO2 nano-branches.
3.3. Catalytic oxidation tests
Fig. 7a shows the degree of discoloration as a function of the reaction time in different catalytic systems. The degree of discoloration is limited to about 20% over a long time, by relying on either MnO2 or H2O2. This indicates a slower oxidation rate or a lower adsorption capacity of MB. For pure GO, 10 min is needed to reach adsorption equilibrium and the degree of discoloration is about 50.2% under the circumstances. If MnO2 and H2O2 coexist in the reaction system, the value of 67.0% is achieved within 1 min. It further increases to a stable value of about 80% after 5 min. However, by replacing them with MnO2/GO-75 composites, the degree of discoloration approaches to around 86.2% within 1 min and at around 99.5% in 30 min. Furthermore, when MnO2/GO-94 composites are used, this value increases to 96.2% within 1 min and only in 10 min, the value approaches to 100%. It is important to note that when MnO2/GO-33 composites were used, the degree of discoloration is lower than that of the other composites. Surprisingly, it was even lower than that of the pure MnO2 system with the assistance of H2O2. Interestingly, after 30 min, the degree of discoloration continues to rise and transcends that of the MnO2 system (87.8%) and reaches above 90%.
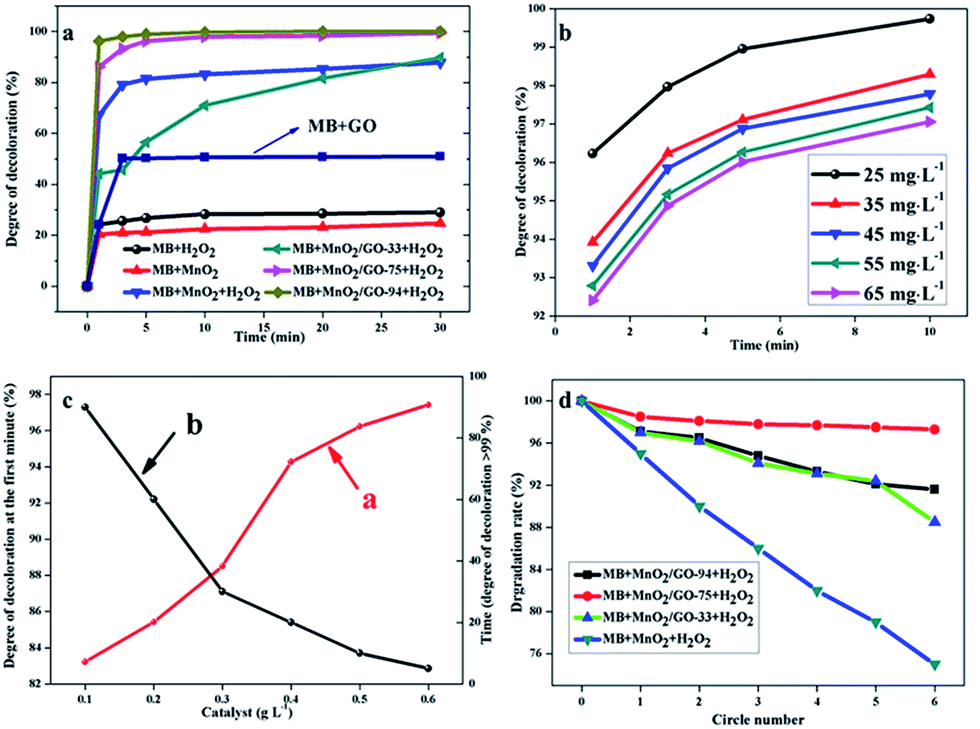 | ||
| Fig. 7 (a) Time profiles of MB discoloration by different catalytic systems under the same reaction conditions; (b) time profiles of MB discoloration by the MnO2/GO-94 catalytic system with different initial concentrations of MB. Reaction conditions: [H2O2]/[MB] = 20 mL L−1, [catalyst] = 0.5 g L−1, T = 25 °C; (c) effect of catalyst amount on the reaction, line (a) (red line) represents the degree of discoloration at the first time interval and line (b) (black line) represents the time value needed when the degree of discoloration is >99%. Reaction conditions: [MB] = 25 mg L−1, [H2O2]/[MB] = 20 mL L−1, T = 25 °C; (d) cyclic tests for catalytic oxidation of MB by different catalytic systems. Reaction conditions: [MB] = 25 mg L−1, [H2O2]/[MB] = 20 mL L−1, [catalyst] = 0.5 g L−1, T = 25 °C. | ||
Hence, the component of β-MnO2 nano-branched–graphene composites has a tremendous impact on the catalytic oxidation activity. Therefore, it is necessary to compare the degree of discoloration at the fifth minute and to compare the time values when the degree of discoloration is greater than 99.5% in different catalytic systems. Table 2 shows that the degree of MB discoloration within 5 min decreases in the following order: MnO2/GO-94 > MnO2/GO-75 > MnO2 > MnO2/GO-33. If we investigate the time value when the degree of discoloration is greater than 99.5%, the order changes to: MnO2/GO-94 < MnO2/GO-75 < MnO2/GO-33 < MnO2.
| Catalyst | Degree of discoloration (%) | Time needed (min) |
|---|---|---|
| MnO2 | 81.44 | 300 |
| MnO2/GO-33 | 56.53 | 130 |
| MnO2/GO-75 | 96.22 | 30 |
| MnO2/GO-94 | 98.77 | 10 |
When the ratio of graphene in β-MnO2 nano-branched–graphene composites increases, the time needed for physical adsorption also increases. Therefore, in a MnO2/GO-33 system, the degree of discoloration is relatively lower at the fifth minute. Once adsorption equilibrium is attained, catalytic reaction becomes faster. The time value when the degree of discoloration is greater than 99.5% in MnO2/GO-33 system is 130 min, which is much shorter than that in pure MnO2 system (300 min). As mass fractions of MnO2 in composites increase, the catalytic reactions become more efficient. These experimental results confirmed that catalytic reactions were mainly initiated by β-MnO2 nano-branches and graphene flakes were used to enlarge adsorption capacity. That is to say, graphene flakes act as the adsorbent and β-MnO2 nano-branches as the catalyst primarily.
Nonetheless, in the pure MnO2 or graphene systems, the degrees of discoloration are much lower. This proves that the synergy effect certainly existed in the β-MnO2 nano-branched–graphene composites, integrating larger adsorption capacity with higher catalytic efficiency.
MB solutions with various initial concentrations of 25–65 mg L−1 were used to carry out catalytic reactions with the same conditions to investigate the effect of initial concentration. The results are shown in Fig. 7b. Increasing the initial concentration reduces the degree of discoloration at a specific time with an unchanged tendency. This is due to the existence of physical adsorption equilibrium. Even in 65 mg L−1 MB solution, the degree of discoloration reaches 97.06% within 10 min. It shows that the new adsorbent–catalyst system in the β-MnO2 nano-branched–graphene composites may have a broader scope of future applications.
To meet the requirements of industrialization, the effect of catalyst input is also studied. The degree of discoloration at the first time (Fig. 7c, line (a), red line) and the time value when the degree of discoloration reaches >99% (Fig. 7c, line (b), black line) were used as key parameters to evaluate the catalytic reactions. Different mass fractions of MnO2/GO-94 were added into MB solution in the presence of H2O2. Changes in the line slopes were monitored and it was shown that to achieve a satisfactory result, higher amounts of catalyst is needed. Nonetheless, the influence of catalyst amount decreases as the amount of catalyst increases. Therefore, an optimal condition of 0.4 g L−1 is chosen, where the degree of discoloration at the first time is 94.27% and only 20 min is needed to reach above 99%.
3.4. Cyclic stability tests
Cyclic tests were used to confirm chemical stability of the as-synthesized β-MnO2 nano-branched–graphene composites (Fig. 7d). By comparing the degradation rates of different catalytic systems, the MnO2/GO-75 composites show the best stability than others after 5 circles, which are attributed to its more stable structure. Although the MnO2/GO-94 composites show a higher catalytic oxidation activity than that of the MnO2/GO-75 composites, the cyclic stability is weaker with a degradation rate of 88.5% after 5 circles. The reason is that massive MnO2 nano-branches are difficult to be dispersed by a spot of graphene flakes. Therefore, after reactions, MnO2 nano-branches become pulverizing and stacking (Fig. S4, ESI†), which has a large effect on catalytic activity. Besides, structures and components of MnO2 remain unchanged, confirmed by XRD and XPS (Fig. S2 and S3 ESI†). Certainly, for pure MnO2 nano-branches, the rate decreased sharply and only 75% are left after cyclic tests. The results prove the novel β-MnO2 nano-branched–graphene composites show enhanced catalytic performances, which are promising for wide practical applications.3.5. Products analysis after reaction
To further define the relation between the degree of discoloration and catalytic oxidation steps, products analysis was conducted in the following work.At first, UV-vis spectroscopy on MB solution was performed at a specific reaction interval (Fig. S5, ESI†). In 5 min, the intensity of two characteristic peaks at 614 nm and 664 nm decreased rapidly. These peaks disappeared as the solution became colorless.
Then, the samples of various time intervals were analyzed by ESI-MS and the results are shown in Fig. 8. The catalytic oxidation process was originated from the breaking of chemical bonds. Before decomposition, MB cations were detected with a strong peak located at m/z = 284 amu (Fig. 8a). Only after 1 min, more peaks appeared, suggesting that the reactions were in progress (Fig. 8b). New peaks at m/z = 270, 256, 242 and 227 amu are potentially due to a N-demethylation oxidative process.42 Other peaks at m/z = 174, 164, 153, 125, 121, 109 and 105 amu are attributed to the further oxidation of aromatic rings during the oxidation process.43 After 10 min, no peak at m/z = 284 amu was detected, showing that MB was completely oxidized into smaller fragments (Fig. 8c). Based on these peaks, the molecular weight of the fragments is calculated to be lower than 196, suggesting that the oxidative process had been effective. The number of peaks in the spectra taken after 30 min was less than those in 10 min (Fig. 8d). It proved that intermediates were further oxidized into smaller molecules.
 | ||
| Fig. 8 ESI-MS analysis on the solution during the reaction process after (a) 0 min, (b) 1 min, (c) 10 min, (d) 30 min. Reaction conditions: [MB] = 65 mg L−1, [H2O2]/[MB] = 20 mL L−1, [Catalyst] = 0.5 g L−1, T = 25 °C. | ||
Finally, inorganic ions, which were possibly generated during the process, such as SO42−, NO3−, Cl−, were detected by ion chromatography. The concentrations of SO42−, NO3−, Cl− in solution after 30 min were of 5.36 μg mL−1, 11.88 μg mL−1 and 16.38 μg mL−1, respectively. These results suggest that the sulfur and nitrogen atoms in MB molecule were completely oxidized into inorganic ions.
4. Conclusion
Novel β-MnO2 nano-branched–graphene composites were fabricated by a controllable ultrasonic-assisted self-assembly process. The β-MnO2 nano-branches were interwoven with graphene flakes to form a stable structure. The novel composites show enhanced catalytic activity and good cyclic stability in catalytic oxidation of MB model. The synergistic effect of combining adsorption efficiency and catalytic efficiency is outstanding. The designed process is cost-effective, easy for scale-up and of mild reaction conditions. These will provide the potential applications in catalytic oxidation of other aromatic compounds. Besides, it also offers new design ideas for the synthesis of graphene-based composites.Acknowledgements
Thanks to the teachers at Materials and Engineering Laboratory (Changchun University of Technology, Changchun 130012, P R China.) and Prof. X. J. Li (Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130012, P R China.) for sample tests.Notes and references
- B. H. Hameed, A. A. Ahmad and N. Aziz, Chem. Eng. J., 2007, 133, 195 CrossRef CAS PubMed.
- F. I. Hai, K. Yamamoto and K. Fukushi, Crit. Rev. Environ. Sci. Technol., 2007, 37, 315 CrossRef CAS.
- J. H. Mo, Y. H. Lee, J. Kim, J. Y. Jeong and J. Jegal, Dyes Pigm., 2008, 76, 429 CrossRef CAS PubMed.
- R. Gong, M. Li, C. Yang, Y. Z. Sun and J. Chen, J. Hazard. Mater., 2005, 121, 247 CrossRef CAS PubMed.
- T. K. Sen, S. Afroze and H. M. Ang, Water, Air, Soil Pollut., 2011, 218, 499 CrossRef CAS.
- S. S. Barton, Carbon, 1987, 25, 343 CrossRef CAS.
- M. Bielska and J. Szymanowski, Water Res., 2006, 40, 1027 CrossRef CAS PubMed.
- V. K. Gupta, D. Pathania, P. Singh, A. Kumar and B. S. Rathore, Carbohydr. Polym., 2014, 101, 684 CrossRef CAS PubMed.
- S. Kant, D. Pathania, P. Singh, P. Dhiman and A. Kumar, Appl. Catal., B, 2014, 145, 340 CrossRef PubMed.
- R. Liang, A. Hu, M. Hatat-Fraile and N. Zhou, Development of TiO2 Nanowires for Membrane Filtration Applications, Nanotechnology for Water Treatment and Purification, Springer International Publishing, 2014 Search PubMed.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, J. Hazard. Mater., 2010, 177, 70 CrossRef CAS PubMed.
- M. Antonopoulou, E. Evgenidou, D. Lambropoulou and I. Konstantinou, Water Res., 2014, 53, 215 CrossRef CAS PubMed.
- H. Y. Shu and M. C. Chang, Dyes Pigm., 2005, 65, 25 CrossRef CAS PubMed.
- H. Kusic, N. Koprivanac and L. Srsan, J. Photochem. Photobiol., A, 2006, 181, 195 CrossRef CAS PubMed.
- A. H. Gemeay, R. G. Ei-Sharkawy, I. A. Mansour and A. B. Zaki, Appl. Catal., B, 2008, 80, 106 CrossRef CAS PubMed.
- B. S. Souza, F. C. Moreira, M. W. C. Dezotti, V. J. P. Vilar and R. A. R. Boaventura, Catal. Today, 2013, 209, 201 CrossRef CAS PubMed.
- W. Zhang, Z. Yang, X. Wang, Y. Zhang, X. Wen and S. Yang, Catal. Commun., 2006, 7, 408 CrossRef CAS PubMed.
- Y. M. Dong, H. X. Yang, K. He, A. Q. Song and A. M. Zhang, Appl. Catal., B, 2009, 85, 155 CrossRef CAS PubMed.
- E. Saputra, S. Muhammad, H. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. B. Wang, Catal. Commun., 2012, 26, 144 CrossRef CAS PubMed.
- Z. H. Ai, L. Z. Zhang, F. H. Kong, H. Liu, W. T. Xing and J. R. Qiu, Mater. Chem. And Phys., 2008, 111, 162 CrossRef CAS PubMed.
- L. Xu, C. Xu, M. R. Zhao, Y. P. Qiu and G. D. Sheng, Water Res., 2008, 42, 5038 CrossRef CAS PubMed.
- R. J. Watts, J. Sarasa, F. J. Loge and A. L. Teel, J. Environ. Eng., 2005, 131, 158 CrossRef CAS.
- N. Sui, Y. Duan, X. Jiao and D. Chen, J. Phys. Chem. C, 2009, 113, 8560 CAS.
- C. L. Yu, G. Li, L. F. Wei, Q. Z. Fan, Q. Shu and J. C. Yu, Catal. Today, 2014, 224, 154 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2006, 56, 1178 CrossRef PubMed.
- E. V. Iski, E. N. Yitamben, L. Gao and N. P. Guisinger, Adv. Funct. Mater., 2013, 23, 2554 CrossRef CAS.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156 CrossRef CAS PubMed.
- H. Chen, S. X. Zhou, M. Chen and L. M. Wu, J. Mater. Chem., 2012, 22, 25207 RSC.
- J. T. Zhang, J. W. Jiang and X. S. Zhao, J. Phys. Chem. C., 2011, 115, 6448 CAS.
- Z. P. Li, J. Q. Wang, X. H. Liu, S. Liu, J. F. Ou and S. R. Yang, J. Mater. Chem., 2011, 21, 3397 RSC.
- H. Chen, L. F. Hu, Y. Yan, R. C. Chen, M. Chen and L. M. Wu, Adv. Energy Mater., 2013, 3, 1636 CrossRef CAS.
- G. X. Zhao, J. X. Li, X. M. Ren, J. Hu, W. P. Hu and X. K. Wang, RSC Adv., 2013, 3, 12909 RSC.
- J. Y. Qu, L. Shi, C. X. He, F. Gao, B. B. Li, Q. Zhou, H. Hu, G. H. Shao, X. Z. Wang and J. S. Qiu, Carbon, 2014, 66, 485 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- J. Y. Zhu and J. H. He, ACS Appl. Mater. Interfaces, 2012, 4, 1770 CAS.
- S. J. Lee, A. Gavriilidis, Q. A. Pankhurst, A. Kyek, F. E. Wagner, P. C. L. Wong and K. L. Yeung, J. Catal., 2001, 200, 298 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS PubMed.
- Z. B. Lei, F. H. Shi and L. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 1058 CAS.
- M. Zaied, S. Peulon, N. Bellakhal, B. Desmazieres and A. Chausse, Appl. Catal., B, 2011, 101, 441 CrossRef CAS PubMed.
- T. Sriskandakumar, N. Opembe, C. H. Chen, A. Morey, C. Kińgondu and S. L. Suib, J. Phys. Chem. A, 2009, 113, 1523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16676k |
| This journal is © The Royal Society of Chemistry 2015 |