
Prussian blue analogues as heterogeneous catalysts for epoxidation of styrene
Abstract
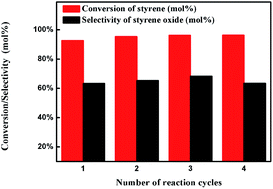
The catalytic epoxidation of styrene was carried out using tert-butyl hydroperoxide (TBHP) in the presence of Prussian blue analogues (PBA) as catalysts and the reaction parameters of the epoxidation, such as temperature, solvent and reaction time, have been optimized. Optimum reaction conditions led to 96% conversion of styrene with selectivity in styrene epoxide equal to 64%. Furthermore, a kinetic investigation of the epoxidation of styrene with TBHP has been studied, a first order with respect to the concentrations of styrene, TBHP and catalyst were determined and an apparent activation energy value of 100.4 kJ mol−1 was obtained.
 Please wait while we load your content...
Please wait while we load your content...

