
DFT study on microstructures and electronic structures of Pt mono-/bi-doped anatase TiO2 (101) surface
Abstract
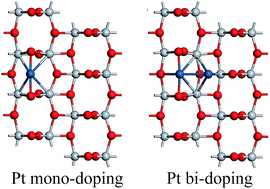
The microstructures and electronic structures of Pt mono- and bi-doped anatase TiO2 (101) surfaces were investigated by density functional theory calculations to elucidate the surface doping effects and the initial stages of Pt cocatalyst formation on a TiO2 photocatalyst surface. Several substitutional and interstitial configurations for the Pt impurity on the surface were studied, and the relative stability of different doping configurations was compared by the impurity formation energy. Under reducing conditions, surface interstitial (in other words, surface adsorbed) doping for Pt is more stable than any competitors (substitutional, bulk or subsurface doping) on an anatase TiO2 (101) surface. Compared with bulk doping, the metal induced gap states were very localized and outstanding in the case of Pt surface interstitial doping onto a TiO2 (101) surface. In the case of Pt substitutional doping, the surface doping effects were harmful for photocatalysis due to the metal induced gap states in the middle of the band gap. However, in the case of Pt interstitial doping, the surface doping effects were very favorable for photocatalysis due to the overlapping of metal induced gap states with the top of the valance band. Systematic calculations revealed that Pt doping is prone to occupy the interstitial sites on the (101) surface. In particular, in the case of Pt bi-doping, two Pt impurity atoms can be co-adsorbed on the surface to form a stable configuration due to the strong Pt–Pt atomic interaction. Therefore, Pt mono-/bi-doping on an anatase TiO2 (101) surface can be considered as the initial stage (nucleation process) of Pt cocatalyst loading onto a TiO2 photocatalyst surface. Therefore, the calculated results can form the basis for further investigations about Ptn cluster loadings or Pt/TiO2 interface formation as well as water molecule adsorption or decomposition on a Pt/TiO2 composite photocatalyst.
 Please wait while we load your content...
Please wait while we load your content...

