
Synthesis of hybrid polymer blend nanoparticles and incorporation into in situ gel foam spray for controlled release therapy using a versatile synthetic purine nucleoside analogue antiviral drug†
Durai Ramyadevi* and
Kalpoondi Sekar Rajan
School of Chemical and Biotechnology (SCBT), SASTRA University, Thanjavur – 613401, India. E-mail: ramya@scbt.sastra.edu; Fax: +91 4362 264120; Tel: +91 9843 361266
First published on 14th January 2015
Abstract
Antiviral therapy requires desired intensity and duration of drug action at the site of infection to minimize adverse effects. The study aims to synthesize a nanoparticle based controlled release system for Acyclovir with required loading and maintenance doses to achieve steady-state plasma concentration for once-daily therapy. The amount of Acyclovir required for 24 h therapy was calculated to be 100 mg, which is approximately 10 fold reduced oral dose. Accordingly, nanoparticles with spherical and smooth surface were synthesized using hybrid polymer blends of polyvinyl pyrrolidone with ethyl cellulose or eudragit RSPO. Physical and chemical interactions between the drug and polymers favoured the formation of stable nanoparticles. Optimized nanoparticles with 80% entrapment exhibited size and surface charge of 100 nm and +26.1 mV respectively. In situ gels incorporated with 20% free drug and 80% Acyclovir loaded nanoparticles showed immediate sol–gel transition at 35 °C–37 °C with gel viscosity ranging between 1000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mPa s. The expected pH independent drug release profile was obtained in 0.1 N HCl, phosphate buffer, simulated vaginal and tear fluids. Ex vivo permeability was significantly higher through cornea than stomach and rectal membranes. Uptake of nanoparticles into human corneal epithelial cell lines was spontaneous. Foam spray was developed for optimized in situ gels for site specific action through vaginal or rectal administration. The bubble size, relative foam density, collapse time, drug content per puff and stability of the foam spray formulations proved its suitability for effective drug delivery. Hence, a unique dosage form achieving predicted release profile through diverse routes was well demonstrated.
000 mPa s. The expected pH independent drug release profile was obtained in 0.1 N HCl, phosphate buffer, simulated vaginal and tear fluids. Ex vivo permeability was significantly higher through cornea than stomach and rectal membranes. Uptake of nanoparticles into human corneal epithelial cell lines was spontaneous. Foam spray was developed for optimized in situ gels for site specific action through vaginal or rectal administration. The bubble size, relative foam density, collapse time, drug content per puff and stability of the foam spray formulations proved its suitability for effective drug delivery. Hence, a unique dosage form achieving predicted release profile through diverse routes was well demonstrated.
Introduction
Acyclovir is a guanosine analogue with the IUPAC name 2-amino-1,9-dihydro-9-((2-hydroxyethoxy) methyl)-6H-purin-6-one, and is identified as one of the potent antiviral drugs for the treatment of herpes simplex virus (HSV-1 and HSV-2) and varicella zoster virus (VZV) infections. In-spite of the emergence of several new antiviral drugs in the market, the older drug Acyclovir is still recommended for clinical application due to its potential capability to decrease viral load, preventing the recurrence of infections and hence remains superior in antiviral therapy. Acyclovir is more effective and well tolerated, which decreases the frequency and severity of asymptomatic HSV-2 reactivation, especially in HIV infected patients.1 However, its biological half life is low (2.5–3.3 h) and it is poorly soluble and permeable. For the recommended oral administration at doses of 200–800 mg 4–5 times daily, it offers very low oral bioavailability (10–20%), and so alternative choice like topical semisolids are required. Various drug modification approaches in terms of development of pro-drugs, techniques for improving the solubility or permeability and design of different drug delivery systems have been adopted to prolong drug release and improve its therapeutic action. Higher antiviral efficacy for treating ocular infections of herpes simplex virus has been demonstrated for newly synthesized dipeptide pro-drugs and amino acid ester pro-drugs.2,3 Even the profitably marketed pro-drugs like Valacyclovir which requires gut wall enzymes for its in vivo activation, seem to be effective by oral therapy (as tablets) and not well accepted for other routes of administration. Also, due to known drawbacks of oral routes of administration, the bioavailability of this pro-drug is reported to be 50–55% only. On the other hand, drug delivery approaches through implantable system,4 nanosponges,5 polymeric nanosuspensions,6 in situ gels,7,8 nanoparticles,9,10 etc., were also reported by researchers to enhance the potential of the drug. Recently, a performance matrix was developed and studied for generic product in vitro bioequivalence to Acyclovir topical creams with reference product Zovirax®.11 Yet, maintenance of desired level of drug concentration at local site of action and systemic circulation throughout the treatment period remains challenging by these modifications and approaches, which provides an opening for development of better drug delivery system.The HSV replication cycle involves the entry of viral DNA into the host nucleus thereby affecting the normal cells and spreading the infection to the nearby cells. The mode of action of Acyclovir is through competitive inhibition of herpes simplex viral DNA polymerase, which occurs most significantly in the infected cells. It is clearly known that Acyclovir is activated by the viral thymidine kinase (TK) and HSV DNA polymerase to convert into Acyclovir triphosphate for its efficient action,12,13 as depicted in Fig. 1. Hence, its role is predominant inside the viral infected cells and remains unused in the normal cells. Moreover, the action of viral TK is 3000 times more effective than host cellular TK and the viral DNA polymerase exhibits 10 to 30 fold greater affinity for Acyclovir phosphate than the host DNA polymerase.14
 | ||
| Fig. 1 Mode of Acyclovir action in HSV infected cells. | ||
The rapid biotransformation of Acyclovir reduces its bioavailability (<20%) and hence >80% of the orally administered doses are excreted as unchanged drug. Saturation kinetics in absorption process is also reported, wherein an increase in dose from 200 to 800 mg decreased the absorption, and therefore reduced the bioavailability from 20 to 10%.14,15 Although Acyclovir is well tolerated in human at regular doses, adverse reactions like nausea, head ache, mild gastro intestinal tract side effects, nephrotoxicity leading to acute renal failure, neurotoxicity including tremors, delirium and seizures have been reported with patients with continuous treatment and over dose.16,17 HSV predominantly affects the mucous tissues in genitals, rectal, nasal, ocular and oral tract and hence higher drug concentration and better local action are essential at these sites. The required dosage of drug should be delivered at controlled rate for longer duration to achieve better pharmacological action. The fact that excretion phenomenon of Acyclovir is biphasic in nature14,18 which has led to failure of many drug delivery systems to maintain the therapeutic level of the drug at required site of action. Hence, achieving the steady state level of drug throughout the duration of therapy has been one of the serious issues for the systems.
Controlled drug release could be achieved through various novel drug delivery systems such as hydrogel, mucoadhesive systems, implants, gelling systems, particulate systems, reservoir devices, etc., depending upon the drug to be loaded and desired release rate.19,20 In situ gelling system loaded with polymeric nanoparticles have been reported recently to achieve controlled release of drugs through ocular route.21–23 In this present work, novel hybrid polymer blend nanoparticles loaded with Acyclovir was first synthesized and then incorporated into a thermo sensitive in situ gelling system. Required amount of burst release of drug to initiate the action followed by controlled release throughout a day was predetermined with its lowest possible dose. The pH-independent biocompatible polymers were used to develop the nanoparticles for effective local and systemic delivery of the drug. The nanoparticles were prepared using hybrid polymer blend combination with varying ratio of amphiphilic and hydrophobic polymers in order to achieve desired entrapped and free drug levels to provide controlled and burst release, respectively. The gel system is designed using poloxamer as gelling polymer as it exhibits remarkable thermo gelling property24,25 through different routes like oral, vaginal, rectal, topical and parenteral (intramuscular). Further, the optimized sol–gel system was converted into foam spray formulation for effective application through vaginal or rectal routes specifically. Foam spray could ensure high level of spreading, local drug delivery with high contact time, improved patient compliance through its ease of application.26,27 The motivation for this study stems from our desire to increase the retention time of drug at target site and provide controlled release as well. A unique drug delivery system suitable for multiple routes with dosing flexibility based on the requirement of the individual patient (individualized therapy) can also be accomplished with this approach.
Materials and methods
Materials
Acyclovir was obtained as gift sample from Matrix India Pvt. Ltd., Hyderabad, India. Eudragit RSPO (ERSPO) was received as gift sample from Glukem Pharma Pvt. Ltd. Hyderabad, India. Poly Vinyl Pyrrolidone K 30 (PVP) and Ethyl Cellulose (EC) were purchased from SD Fine Chem. Pvt. Ltd., Mumbai, India. Pluronic F-127 (PF-127) was procured from Sigma Aldrich, Mumbai, India. All other chemicals and buffer reagents used in the study were of analytical grade.Pharmacokinetics for controlled release of Acyclovir
The dose required to provide controlled release of Acyclovir at target site can be predicted from its pharmacokinetic profile using one compartment model. It has been shown in clinical trials that 200 mg dose of Acyclovir every 4 h for 6 times a day yielded the steady state drug level on the 2nd day only. The mean plasma concentration with trough and peak levels was 0.31 and 0.49 μg mL−1 respectively.14,28 Multiple oral dosage regimens require more than 6 half lives to attain the steady state drug level in plasma.The total amount of drug required in the body to achieve the plasma concentration can be calculated from the expression,
 | (1) |
 | (2) |
| K = Di × Ke | (3) |
| Dm = K × T | (4) |
| Dl = Di − (K × Tp) | (5) |
| Dt = Dl + Dm | (6) |
Preparation of nanosuspension
Nanoprecipitation method30 was adopted to prepare the hybrid polymer blend nanoparticles and study the effect of varying composition of amphiphilic polymer (PVP) and hydrophobic polymers (EC and ERSPO) on the entrapment efficiency of Acyclovir. Typical trial experiments were performed with hybrid polymer blends to identify conditions to achieve 80% entrapment of the drug. The polymers were dissolved in 5 mL of methanol and 50 mg of the drug was dissolved in 10 mL of aqueous surfactant solution (1% w/v or 0.5% w/v Pluronic F-127). The organic phase was transferred into aqueous phase drop-wise with moderate magnetic stirring at room temperature, until the organic solvent evaporated completely with spontaneous formation of the nanoparticles.The formulated nanosuspension was centrifuged using cooling ultracentrifuge (Sigma 3K30, Osterode Am Harz, Germany) at 16000 rpm for 30 min at −4 °C, and the nanoparticles were pelletized. This pellet was re-dispersed in distilled water using cyclone mixer (Remi CM101 DX, Mumbai, India) to obtain uniform nanoparticle dispersion, which was freeze-dried (Christ Alpha 2-4 LD Plus, Osterode Am Harz, Germany) and used for further studies.
Entrapment efficiency of nanoparticles
The supernatant solution obtained after ultracentrifugation was suitably diluted and the absorbance of the samples were measured using UV-visible spectrophotometer (Evolution 201, ThermoScientific, USA) at λmax of 252 nm. The concentration of Acyclovir was estimated using the calibration standard curve and the entrapment efficiency was calculated using the formula,31
 | (7) |
All experiments were repeated thrice and results have been reported as mean ± standard deviation.
Characterization of the nanoparticles
The size distribution and surface charge of the drug loaded nanoparticles were measured using Zeta Sizer (Nano Series ZS, Malvern, UK) to assess the distribution and stability of the formulation. The surface morphology of the nanoparticles was observed using field emission scanning electron microscope (JSM 6701F, JEOL, Japan) and compared with the pure drug. The freeze-dried samples were sputter coated with gold using auto fine coater (JFC 1600, JEOL, Japan), and then placed in the sample holder for imaging. The morphology of the nanoparticles dispersed in different media was analyzed to assess the possible correlation in drug release studies. Pure drug, pure polymers and the synthesized nanoparticles were subjected to FTIR spectroscopy (Perkin-Elmer System 200, Shelton, Connecticut, USA) to identify the interactions between them. After uniform blending with dried IR grade potassium bromide crystals and samples were pelletized into thin disc using hydraulic pellet press (Kimaya Engineers, Thane, India) at 60 kg cm−2 and IR spectrum was recorded between wave number ranging 4000–400 cm−1.Preparation of thermosensitive gelling system
Cold method32 was adopted with slight modification for the preparation of thermosensitive sol–gel systems using different poloxamer concentrations (12–24% w/v), in addition with 0.01% benzalkonium chloride as preservative. The required amount of polymer was gradually dispersed in neutral aqueous solution with gentle stirring and kept at 4 °C for about 24 h, until complete solubilisation of the polymer to form homogenous sol. The required mass of polymeric nanoparticles and the free drug, equivalent to 80% and 20% of requisite dose of Acyclovir respectively, were added to the sol. The samples were mixed uniformly to obtain a final drug concentration of 10 mg mL−1 and then refrigerated for further research. The formulations were sterilized with UV radiation before sterility testing.Estimation of pH and gelation
The pH of the samples was measured in triplicates using electronic pH meter (pHep, Hanna Instruments, Woonsocket, Rhode Island, USA), and the results expressed as mean ± SD. Experiment to assess the critical gelation temperature (CGT) for sol to gel transition was carried out through modified Miller and Donavan technique.33 About 2 mL aliquot of sol was transferred to test tubes, immersed in a water bath at 4 °C and sealed with aluminium foil. The temperature of water bath was increased in increments of 1 °C and left to equilibrate for 2 min at each new setting. Critical gelation temperature was noted when the meniscus of sample remained without flowing upon tilting through 90°.34Drug content analysis
About 1 mL of the sample was dissolved in 100 mL of distilled water and kept overnight for complete release of the drug. The absorbance of the solution was measured using UV-visible spectrophotometer at 252 nm using water as blank. The experiment was performed in triplicates for each formulation and the amount of Acyclovir was calculated using standard calibration curve.Rheology studies
Viscosity of the formulations was determined using Brookfield Viscometer (Brookfield Viscometer DV-II + Pro Extra, USA) with spindle no. 63. Viscosity of the sol (before gelling) was measured at room temperature (25 °C), at varying shear rate. The temperature of the sol was progressively increased from 25 °C to 37 °C at rate of 2 °C min−1 and the viscosity of the sample was recorded at same shear rates after gelling of the sample. All measurements were recorded in triplicate with good reproducibility.In vitro release studies
In vitro release of Acyclovir from the thermo sensitive in situ gel formulations was studied for the period of 24 hours by dialysis bag method using 4 different media. Phosphate buffer of pH 7.4 and 0.1 N HCl pH 1.2 (without enzymes) were used to mimic the systemic circulation and gastric (stomach) environment, respectively. The simulated vaginal fluid (pH ∼ 4.2) and simulated tear fluid (pH ∼ 7.4) resemble the biological media in vaginal and ocular route of administration.The release profile of the optimized hybrid blend polymeric nanoparticles loaded with Acyclovir (AN-1 and AN-2) was studied before its incorporation into the gelling system. Similarly the drug release pattern from thermo sensitive in situ gel incorporated with plain Acyclovir (AIG-1 to AIG-5) was also evaluated. The comparative study was performed to ascertain the efficiency and significance of the dual controlled delivery system (polymeric nanoparticles + thermo sensitive gel system).
About 1 mL of the prepared sol system was placed in a dialysis bag (HiMedia, Mumbai, India) and immersed in 5 mL of media in a vial with a small magnetic bead at the bottom. The vial kept on a water bath and the entire set up was positioned on a magnetic stirrer (100 ± 5 rpm) and maintained at temperature of 37 °C ± 2 °C. At periodic time intervals entire media was withdrawn from the vial and replaced with same volume of warm fresh media. The collected samples were analyzed using UV-visible spectrophotometer at λmax of 242 nm for estimation of drug release from the gel. The experiments were performed in triplicates and data presented as the mean with standard deviation.
Kinetics of drug release
The mechanism of drug release from the in situ gelling system was predicted through the drug release kinetics, by fitting the drug release data to various models such as zero-order, first-order, Higuchi, Hixson, Korsmeyer–Peppas, Hopfenberg, Baker–Lonsdale, Makoid–Banakar, Weibull and Gompertz models. The values of R2, n (release exponent), K (rate constant) and SSR (Sum of Squared Residual) were computed for each formulation and the possible mechanism of drug release was identified.Sterility studies
These tests were done to detect the presence of viable forms of bacteria, fungi and yeast in the formulations. The sol samples were added individually into the plates containing nutrient agar and incubated at ambient conditions for 48 hours, and then the plates were examined. The procedure was carried out under laminar air flow sterile area and the experiments adhered to circumvent accidental contamination of the product during the test.35Stability studies
The optimized thermo sensitive gelling systems that satisfied the expected physico-chemical standards and drug release pattern were evaluated for their stability on storage. Stability studies were conducted as real time tests at ambient room temperature (25–27 °C) and at refrigerated temperature (2–8 °C). Samples were withdrawn from the stored formulations at weekly intervals and evaluated for its appearance, pH, drug content, viscosity and gelling property.Ex vivo permeation study
The Franz diffusion apparatus (in-house made) with effective diffusion area of 2.54 cm2 was used to determine the amount of drug diffused across different ex vivo membranes. The biological membranes such as rectal membrane, stomach membrane and corneal membrane were isolated from goat tissue obtained from local slaughter house. The receptor cell was filled with phosphate buffer pH 7.4 as medium, till the neck of the chamber (15 mL) and the excised membrane was mounted between donor and the receptor. The in situ gel formulation was placed on the upper surface of membrane, while the lower surface was in contact with the medium of the receptor cell. The entire apparatus was kept over magnetic stirrer with a magnetic Teflon bead placed in the bottom of the receptor cell. Samples of 5 mL were collected from the receptor cell at periodic time intervals, and replaced with the same volume of fresh media to maintain perfect sink condition. The absorbance of collected samples was measured at 252 nm (λmax) of Acyclovir using respective blank. The amount of drug diffused through the membrane at each time point was calculated to assess the cumulative drug diffusion.36Permeation data analysis
The amount of material flowing through a unit cross section of a membrane per unit time is measured as flux, and expressed as,
 | (8) |
The permeation profile was constructed by plotting the cumulative amount of drug permeated per unit surface area of the membrane (μg cm−2) versus time (h), and the slope of linear portion of plot was estimated as steady state flux (Jss, μg cm−2 h−1). The lag time (tL, h) was estimated from the x-intercept of the slope and the permeability coefficient (Kp, cm h−1) was calculated by using the eqn (9).
 | (9) |
Statistical studies
The statistical analysis was performed through two-way ANOVA at p < 0.05 level of significance for the data obtained by in vitro drug release and ex vivo permeation studies.Cell uptake studies
Human corneal epithelial (HCE) cells obtained from Sankara Nethralaya Eye Hospital, India were cultured in Dulbecco's Modified Eagles Medium/F12 containing 10% fetal bovine serum (FBS), 1% penicillin–streptomycin (P–S) and 1% epithelial growth supplement. The cells were detached to develop single cell suspensions and counted using a haemocytometer. The viable cells were seeded on cover slips in 6-well plate at density of 100000 cells per well and incubated for 24 hours at 37 °C in 5% CO2 with 10% FBS. Nanoparticles (AN-1 and AN-2) marked with curcumin fluorescence were individually loaded into the wells at concentration of 50 μg mL−1 and incubated for 30 min to 3 hours. The uptake of nanoparticles by the epithelial cells was viewed using laser scanning confocal microscopy (Olympus FV-1000, Japan) at 60X magnification with the excitation wavelength of curcumin.
To determine that the intracellular fluorescence is not due to uptake of the dye that might have been released from the nanoparticles, a sample of nanoparticles was suspended in cell medium (5 mL) and dialyzed against plain cell medium (5 mL) for 3 h using a dialysis bag.37 The percentage of free curcumin released in the media was measured using UV-visible spectrophotometer with respect to standard calibration curve. The entrapment efficiency of curcumin in the nanoparticles was estimated and hence, the percentage of curcumin available in the nanoparticles after 3 h dialysis in cell media was estimated.
Development of foam spray
In order to facilitate effective and easy application through vaginal and rectal routes, the optimized in situ gelling systems of PEC-3 and PERS-2 were developed into foam spray in 250 mL canisters by compression filling technique. To achieve the foam delivery of the sol system, 20–30% of hydro fluoro carbon (1,1,1,2-tetra fluoro ethane) was used as the propellant.38 The sol sample was placed in the canister followed by crimping the valve assembly and removal of air by vacuum. Propellant in the form of compressed gas was then passed into the canister with 2–3 bars pressure and finally sealed. The canisters were stored in refrigerator to maintain the sol integrity of the formulation to enable easy delivery of the spray upon actuation of the valve.Evaluation of foam spray
The developed foam spray was evaluated for physico chemical characteristics to assess their suitability for application. Foam structure and foam characteristics such as bubble size, shape, clarity and collapse time were depicted by visual examination. Bubble size was observed immediately after spraying out of canister to identify its coarse or fine appearance, followed by the optical microscopic (KI-198A, Khera Instruments Pvt. Ltd., India) examination. The foam structure as well as foam clarity was assessed as it plays a role in aesthetic value. Collapse time was noted as the time taken by the bubbles to disappear completely to leave the formulated drug over the applied surface.39,40 Relative foam density was estimated by dispensing the foam into a flat bottomed dish with approximate volume of 60 mL and measuring its weight. Similarly mass of the same volume of water was determined and the relative foam density was calculated by the formula,27
 | (10) |
Drug content per puff of foam (approximately 10 mL) was determined by suitably diluting the sample and measuring the absorbance using UV-visible spectrophotometer.41 All the experiments were performed in triplicate and the results were tabulated as mean with standard deviation.
Results and discussion
Prediction of controlled drug release profile and reduction of dose of Acyclovir
The amount of Acyclovir required for achieving the mean plasma concentration of 0.31–0.49 μg mL−1 was calculated from its volume of distribution (Vd = 32.4–61.8 L) as follows:| X = Vd × C | (11) |
| Amount of drug in body = (32.4 to 61.8 L) × (0.31 to 0.49 μg mL−1) = 14.6 to 23.07 mg = 20 mg approximately |
For the elimination half life of 2.5 to 3.3 h, the elimination rate constant for Acyclovir is estimated to be 0.238 h−1. As the values of Di and Ke are 20 mg and 0.238 h−1 respectively, the rate constant (K) for controlled release of Acyclovir is found to be 4.76 mg h−1 from eqn (3). The controlled release of Acyclovir for once daily administration for total drug release period (T) of 20 h requires the maintenance dose (Dm) of approximately 95.2 mg, as estimated using eqn (4). Hence, from eqn (5), the loading dose (Dl) required to achieve the desired rate of controlled release for reaching peak plasma concentration time (Tp = 1.5 to 2.5 h) is found as 10.48 mg. So, according to eqn (6), the total dose (Dt) required for the dosage form to achieve the desired amount of drug release is calculated to be 105.68 mg. The expected Acyclovir controlled drug release profile, cumulative amount of drug remaining in body after each half life, steady state plasma concentration at each half life and its biological elimination profile are shown in Fig. 2.
 | ||
| Fig. 2 Predicted controlled release profile of Acyclovir for once daily administration. | ||
The prediction of once-a-day dosage system clearly indicated that the quantity of Acyclovir required for controlled release is 105 mg. This once-a-day dose calculated is approximately 10 times lower than the dosage currently prescribed (200 mg 5 times per day orally). The reduction of dose could ultimately decrease the excess drug exposure and associated adverse side effects and also the controlled release of drug reduces the fluctuations in drug levels in vivo.42 The predicted drug release profile clearly displayed 20% of burst release within 2–3 h (1st half life) to reach the minimum effective concentration (MEC) in plasma, followed by controlled release of remaining 80% of the drug. The release kinetics was found to fit Korsemeyer–Peppas and Hopfenberg models with high correlation coefficient (R2 > 0.99) that explained drug release by diffusion and surface erosion from matrix. Hence a controlled release system that provides required loading dose for initial burst release and maintenance doses for total period of 20 h has been designed.
Optimization of entrapment level in nanoparticles
A range of polymer blend combinations were characterized to identify the formulation with required encapsulation efficiency. Entrapment efficiency was found to increase with increase in amount of polymer used for nanoparticle formulation. However very high level of the polymer caused reduction in entrapment (results not shown) due to rapid precipitation and sedimentation of excess polymer during the preparation.43The entrapment efficiency of Acyclovir was found to be 80 ± 2% in two formulations, AN-1 and AN-2. The formulation AN-1 was prepared using blended polymers PVP and EC at the concentrations of 20 mg mL−1 and 10 mg mL−1 respectively, while 1% w/v Pluronic F-127 was used as surfactant. Whereas formulation AN-2 was developed using the combination of polymers PVP (10 mg mL−1) and ERSPO (5 mg mL−1) with 0.5% w/v Pluronic F-127 surfactant.
As the system was designed to accomplish controlled release of 80% drug for longer time, the initial stage of optimization was aimed for 80% entrapment of the drug in the polymeric nanoparticles. Hence, formulations which exhibited encapsulation efficiency of 80 ± 2% were selected for further characterization, followed by incorporation into the thermo sensitive gelling system.
Size and surface charge of nanoparticles
The average diameter of the nanoparticles in formulation AN-1 and AN-2 were found to be 400 nm and 100 nm, respectively, which could be one of the key factors for effective drug delivery to improve bioavailability.44 The SEM images (Fig. 3a–c) of pure drug compared to the formulated nanoparticles, confirmed the transition of non uniform flakes of Acyclovir into uniform spherical and smooth particles. Both the formulations exhibited polydispersity in size with wide particle size range from 310 nm to 540 nm and 45 nm to 255 nm for AN-1 and AN-2, respectively. | ||
| Fig. 3 SEM images of (a) pure drug (b) Acyclovir loaded PVP–EC nanoparticles (c) Acyclovir loaded PVP–ERSPO nanoparticles. | ||
The lyophilized nanoparticles in different release media showed fused spherical particles45,46 (Fig. 4). This could be due to the hydrolysis and diffusion of amphiphilic PVP molecules from nanoparticle matrix to the external surface in presence of the media. Yet, the spherical nature of the particles without significant changes in size and its wide distribution could be observed. The fusion of nanoparticles was more in the PVP–EC blend nanoparticles compared to the PVP–ERSPO system, which could be attributed to the presence of higher amount of PVP in the former.
 | ||
| Fig. 4 Morphology of nanoparticles in different media (A) AN-1 nanoparticles in 0.1 N HCl (B) phosphate buffer (C) simulated vaginal fluid (D) simulated tear fluid (E) AN-2 nanoparticles in 0.1 N HCl (F) phosphate buffer (G) simulated vaginal fluid (H) simulated tear fluid. | ||
The zeta potential was −12.3 mV and +26.1 mV for the formulations AN-1 and AN-2 respectively. Surface charge of nanoparticles was influenced by the type of polymer (anionic or cationic), therefore EC incorporated nanoparticles showed negative charge and ERSPO system exhibited positive charge. Also the zeta potential was found to be independent of the amount of PVP in both systems due to its amphiphilic non-ionic nature. It is known that as the cellular membranes are negatively charged, cationic particles exhibited higher tendency to permeate membranes thereby improving the cellular uptake of nanoparticles.47
Interaction of drug and polymers in nanoparticles
FTIR spectrum of the drug loaded nanoparticles compared with the pure drug and pure polymers is displayed in Fig. 5. Acyclovir identity was confirmed by the presence of characteristic peaks at 3440 cm−1 for alcoholic –OH group, 3182 cm−1 and 2854 cm−1 for –CH stretching, 1714 cm−1 for –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ketone stretching, 1641 cm−1 for –CC stretching, 1387 cm−1 for presence of amine group, 1105 cm−1 for –C–N stretching and 902 cm−1for –N–H stretching. Pure polymers PVP, EC and ERSPO showed their characteristic bands for their identity.
O ketone stretching, 1641 cm−1 for –CC stretching, 1387 cm−1 for presence of amine group, 1105 cm−1 for –C–N stretching and 902 cm−1for –N–H stretching. Pure polymers PVP, EC and ERSPO showed their characteristic bands for their identity.
 | ||
| Fig. 5 Chemical interaction studies of drug and polymer by FTIR analysis. | ||
Acyclovir loaded PVP–EC blend nanoparticles showed the bands of drug at slightly shifted wave numbers such as 3437 cm−1 for –OH stretching, 2974 cm−1 for aromatic –C–H stretching, 1663 cm−1 for –CO for ketone stretching, 1384 cm−1 due to amine group and 1109 cm−1for –C–N stretching. In case of Acyclovir loaded PVP–ERSPO blend nanoparticles, the bands of drug were observed at 3437 cm−1, 1651 cm−1, 1385 cm−1 and 1106 cm−1. Shift and broadening of bands in the nanoparticles could be due to overlapping of peaks of similar functional groups –CO and –C–H and –N–H in both drug and polymers, hydrophobic interactions and existence of hydrogen bonding interactions due to free hydroxyl and carbonyl groups in Acyclovir and PVP, respectively.48,49 However, uniqueness of drug was not affected by these interactions, as confirmed by the presence of its characteristic peaks.50,51
Characteristics of the in situ gels
The results of characterization studies of formulated Pluronic in situ gels, PEC-1 to PEC-5 and PERS-1 to PERS-5 containing AN-1 nanoparticles and AN-2 nanoparticles, respectively are shown in Table 1. Physical appearance of the sol systems showed that transparency and clarity decreased with increase in the polymer content, especially at 24% w/v concentration of Pluronic F-127. However, the pH and drug content lied within the expected range of 6.8 ± 0.5 to 7.0 ± 0.3 and 97.1 ± 5.8 to 101.8 ± 3.2%, respectively. The sols converted into gel spontaneously at the critical gelation temperature of 35–37 °C in between 20–50 seconds.| Tests | PEC-1 | PEC-2 | PEC-3 | PEC-4 | PEC-5 | PERS-1 | PERS-2 | PERS-3 | PERS-4 | PERS-5 |
|---|---|---|---|---|---|---|---|---|---|---|
| Pluronic F-127 (% w/v) | 12 | 15 | 18 | 21 | 24 | 12 | 15 | 18 | 21 | 24 |
| Clarity | Clear | Clear | Clear | Slightly turbid | Turbid | Clear | Clear | Clear | Slightly turbid | Turbid |
| pH | 6.9 ± 0.1 | 7.03 ± 0.05 | 7 ± 0.2 | 6.96 ± 0.2 | 7 ± 0.3 | 6.9 ± 0.1 | 6.86 ± 0.05 | 6.93 ± 0.2 | 6.96 ± 0.2 | 6.9 ± 0.17 |
| Gelling temp. | 37 °C | 37 °C | 36 °C | 37 °C | 35 °C | 37 °C | 37 °C | 35 °C | 37 °C | 36 °C |
| Gelling time (sec.) | 47 ± 2.6 | 40 ± 2.5 | 30 ± 1.1 | 24 ± 2 | 25 ± 1 | 45 ± 2.5 | 32 ± 2.5 | 23 ± 1 | 24 ± 2 | 28 ± 2 |
| Drug content (%) | 100.6 ± 2.1 | 98.5 ± 0.8 | 99.9 ± 1.4 | 98.9 ± 6.1 | 99.6 ± 4.8 | 97.1 ± 5.8 | 100.6 ± 3.3 | 100.9 ± 4.7 | 101.8 ± 3.2 | 98.7 ± 4.1 |
Impact of rheological behaviour
The viscosity measurements obtained at room temperature (25 °C) and body temperature (37 °C) clearly demonstrated the sol to gel transition at its critical gelation temperature. The viscosity-shear rate data for the formulated in situ gels (PEC-1 to PEC-5 and PERS-1 to PERS-5) showed shear thinning characteristics at both sol and gel state (Fig. 6a–d). Greater the concentration of Pluronic F-127, higher is the viscosity of the formulations. The viscosity of PEC formulations containing 12–24% w/v concentration of Pluronic varied from 12–52 mPa s at shear rate of 264 s−1, respectively. The PERS formulations also showed analogous behaviour with viscosity ranging 20–106 mPa s. The gel phase of both PEC and PERS systems exhibited viscosity in the range of 1000–10000 mPa s.
 | ||
| Fig. 6 Viscosity of thermo sensitive in situ gels loaded with Acyclovir nanoparticles (a) PEC sol (b) PEC gel (c) PERS sol (d) PERS gel. | ||
The decrease in viscosity with respect to increase in shear rate indicated the non-Newtonian flow behaviour with pseudo plastic nature of Pluronic sol–gel systems. The shear thinning property of the Pluronic gels is attributed to the formation of spherical micelles by the unimers, leading to clusters and hard sphere crystals, which could align under the influence of shear during the measurement of viscosity.34 At low shear rate, the high viscosity of gel could support in maintaining effective contact on the surface applied. Shear thinning character of gel could permit appropriate delivery and distribution of the dose over the surface. The high viscosity range of gel systems could enhance in vivo bioadhesion (mucoadhesion) irrespective of the pH of the environment.52
Various mechanisms have been explained for the sol to gel transition process of this polymer.53,54 Poloxamer being an amphiphilic co-polymer containing the PEO–PPO–PEO block chains, the gel formation was attributed to entropic changes in the system leading to ordered water molecules close to PPO core, by which the monomers aggregated as micelles to minimize the free energy of solution. As the temperature increased further, micelles undergo progressive dehydration causing entanglement by hydrophobic associations, which results in micelle crystallization in a cubic lattice.55
Dual controlled release behaviour
The drug release of the two in situ gelling systems (PEC and PERS) exhibited controlled release behaviour in four different media, and was compared with the predicted controlled release profile of Acyclovir, as shown in Fig. 7a–d and 8a–d. Controlled release of drug was achieved by the formulations through the dual barriers of the hard sphere crystal of micelle packing and the polymeric encapsulation of the nanoparticles. Burst release of the drug was accomplished between 2–3 hours due to the single micelle barrier for the free drug in gelling polymer, which could satisfy the initial loading dose required to achieve minimum therapeutic level in vivo. However, drug release from polymeric nanoparticles incorporated in the gel could be achieved through several mechanisms such as degradation of polymer PVP by hydrolysis, diffusion of drug from polymeric nanoparticles, diffusion of drug through the micellar aggregates of poloxamer, dilution of gel by the media leading to its erosion and dissolution.56 Drug release from this system is also influenced by viscosity of gel, presence of aqueous channels, distribution of free drug and nanoparticles between the micelles. Higher the viscosity of the gelling system, lesser is the drug release.55 | ||
| Fig. 7 Drug release profile of PEC in situ gelling systems in media (a) 0.1 N HCl (b) phosphate buffer (c) simulated vaginal fluid (d) simulated tear fluid. | ||
 | ||
| Fig. 8 Drug release profile of PERS in situ gelling systems in media (a) 0.1 N HCl (b) phosphate buffer (c) simulated vaginal fluid (d) simulated tear fluid. | ||
In case of PEC systems, PEC-1 and PEC-2 showed faster drug release, whereas PEC-4 and PEC-5 displayed very slow release due to low and high concentration of gelling polymer, respectively. PEC-3 containing 18% of PF-127 incorporated with Acyclovir loaded PVP–EC blend hybrid nanoparticles depicted the expected drug release profile. The PERS systems also exhibited similar release pattern, wherein the formulation PERS-2 containing 15% of PF-127 incorporated with Acyclovir loaded PVP–ERSPO nanoparticles matched the expected release profile. As the systems were loaded with equivalent level of the nanoparticles (80%) and free drug (20%), the amount of drug release was found to be dependent on the concentration of gelling polymer.
Acyclovir loaded polymeric nanoparticles AN-1 and AN-2 showed sustained release of drug for a period of 13 h and 24 h, respectively (Fig. 9a). Likewise, in situ gels loaded with unprocessed (pure) Acyclovir (AIG-1 to AIG-5) exhibited faster drug release reaching 100% concentration within 8 h for 12% w/v PF-127, as shown in Fig. 9b. It was extended to the maximum period of 15 h for the 24% w/v concentration of polymer. Also the release profile and kinetics were typically different from the expected pattern. Yet the combination of both techniques (polymeric nanoparticles and in situ gel) provided a dual controlled system, which satisfied the required release behaviour.
 | ||
| Fig. 9 Drug release profile (a) polymeric nanoparticles of Acyclovir (b) plain drug loaded in situ gels. | ||
The usage of different media for the in vitro release studies aided in prediction of the in vivo dissolution through various routes of administration. The morphology of the nanoparticles did not show considerable variation with respect to the different media used in the study. And the drug release phenomenon for the formulations in all the four media was found to be similar, due to pH independent drug release mechanism from the systems. However, considerable variation between the optimized formulations PEC-3 and PERS-2 was observed in-spite of both showing the expected release pattern. This is attributed to higher hydrophobicity of ERSPO due to methacrylic chains57 compared to EC containing cellulose network. Yet, PEC-3 containing higher concentration of gelling polymer (18% w/v) than PERS-2 (15% w/v) offered higher number of micelles for packing into hard sphere crystals that controlled the drug release. The variation in drug release could also be attributed to the presence of high concentration of PVP polymer and surfactant in AN-1 nanoparticle incorporated in PEC systems, compared to the less PVP and surfactant level in AN-2 nanoparticles loaded into the PERS systems. PVP exhibited vital role to enable faster release due to its hydrolysis followed by pore diffusion of drug.58
Mechanism of drug release
The data obtained from the drug release kinetics are shown in Table 2 for the PEC and PERS thermosensitive gelling systems loaded with the Acyclovir hybrid blend polymeric nanoparticles. The release kinetics of formulations was best fitted with Korsemeyer–Peppas kinetics and Makoid–Banakar model with R2 > 0.99 and least SSR values. The mechanism of drug release was based on non-Fickian diffusion transport as indicated by the release exponent n-value >0.5. Further, the R2 > 0.98 with the lowest SSR value in Weibull and Hopfenberg model suggested the drug release from polymeric matrix system through surface erosion of the gel matrix.59 This was due to the interaction of the poloxamer (physical entanglements or hard sphere crystals of micelles) with physiological fluid. The CGC of the sample was decreased after prolonged exposure at physiological condition, finally leading to dissolution and erosion of the gel through its surfaces.60| Kinetic models |
PEC-3 | PERS-2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug release media | HCl | PB | SVF | STF | HCl | PB | SVF | STF | |
| a HCl – 0.1 N HCl pH 1.2, PB – phosphate buffer pH 7.4, SVF – simulated vaginal fluid pH 4.2, STF – simulated tear fluid pH 7.4. | |||||||||
| Zero order | R2 | 0.9273 | 0.9052 | 0.9298 | 0.9 | 0.9194 | 0.9256 | 0.9193 | 0.9212 |
| K0 | 4.72 | 4.76 | 4.73 | 4.88 | 4.7 | 4.64 | 4.66 | 4.59 | |
| SSR | 1059 | 1367 | 1023 | 1496 | 1122 | 1018 | 1095 | 1047 | |
| First order | R2 | 0.9854 | 0.9897 | 0.9844 | 0.9892 | 0.9809 | 0.9804 | 0.9819 | 0.9821 |
| K1 | 0.08 | 0.083 | 0.08 | 0.087 | 0.08 | 0.078 | 0.079 | 0.077 | |
| SSR | 212 | 148 | 227 | 161 | 266 | 267 | 245 | 238 | |
| Higuchi | R2 | 0.9384 | 0.9499 | 0.9392 | 0.9539 | 0.9474 | 0.9446 | 0.9496 | 0.9484 |
| KH | 16.99 | 17.24 | 17.02 | 17.68 | 16.97 | 16.74 | 16.82 | 16.57 | |
| SSR | 896 | 722 | 887 | 690 | 731 | 758 | 684 | 685 | |
| Korsemeyer–Peppas | R2 | 0.9914 | 0.9908 | 0.9927 | 0.9918 | 0.9931 | 0.9935 | 0.9944 | 0.9942 |
| KKP | 10.12 | 11.15 | 10.09 | 11.68 | 10.57 | 10.19 | 10.53 | 10.3 | |
| SSR | 124 | 132 | 105 | 123 | 96 | 88 | 76 | 76 | |
| n | 0.714 | 0.681 | 0.716 | 0.672 | 0.696 | 0.705 | 0.694 | 0.697 | |
| Hixson–Crowell | R2 | 0.9932 | 0.993 | 0.9927 | 0.9929 | 0.987 | 0.9871 | 0.9871 | 0.9866 |
| KHC | 0.023 | 0.024 | 0.023 | 0.025 | 0.023 | 0.023 | 0.023 | 0.022 | |
| SSR | 98 | 100 | 107 | 106 | 180 | 176 | 174 | 177 | |
| Hopfenberg | R2 | 0.9935 | 0.9933 | 0.993 | 0.9931 | 0.9871 | 0.9873 | 0.9871 | 0.9866 |
| KHB | 0.027 | 0.02 | 0.028 | 0.021 | 0.025 | 0.025 | 0.023 | 0.022 | |
| SSR | 94 | 97 | 101 | 103 | 179 | 174 | 174 | 177 | |
| Baker–Lonsdale | R2 | 0.8906 | 0.9053 | 0.8907 | 0.9083 | 0.9006 | 0.8977 | 0.9035 | 0.9033 |
| KBL | 0.006 | 0.007 | 0.006 | 0.007 | 0.006 | 0.006 | 0.006 | 0.006 | |
| SSR | 1593 | 1366 | 1594 | 1372 | 1383 | 1400 | 1311 | 1284 | |
| Makoid–Banakar | R2 | 0.9945 | 0.995 | 0.9949 | 0.9956 | 0.9934 | 0.9937 | 0.9946 | 0.9943 |
| KMB | 8.25 | 9.02 | 8.55 | 9.63 | 9.96 | 9.74 | 10.12 | 9.96 | |
| SSR | 79 | 71 | 74 | 65 | 91 | 86 | 73 | 75 | |
| Weibull | R2 | 0.9953 | 0.9952 | 0.9948 | 0.9947 | 0.9911 | 0.991 | 0.9916 | 0.99 |
| SSR | 67 | 69 | 75 | 78 | 123 | 122 | 114 | 132 | |
| Gompertz | R2 | 0.9369 | 0.9413 | 0.9338 | 0.9389 | 0.9203 | 0.9201 | 0.9211 | 0.9226 |
| SSR | 919 | 846 | 966 | 914 | 1109 | 1092 | 1071 | 1028 | |
Sterility and stability of the systems
Test for sterility is an important aspect for preparations intended for ophthalmic and other mucosal delivery systems since non-sterile formulations could induce and spread pathogenic infections at the site of application. The optimized formulations (PEC-3 and PERS-2) did not exhibit any symptoms of microbial or fungal growth during the tests for its sterility, which confirmed its suitability for in vivo application. Addition of benzalkonium chloride during the preparation and the UV sterilization of the final product maintained the sterility of the dosage form. There was no significant difference in the pH, viscosity and uniformity of drug content of formulations stored at 2–8 °C. Hence, storage at refrigerated condition was more appropriate to maintain the sol integrity of the formulation which could favour easy application to necessary site.Ex vivo permeation studies
The permeation profile and comparative data are shown in Fig. 10a and b and Table 3 for the optimized formulations PEC-3 and PERS-2. The cumulative amount of drug permeated through all the membranes was found to be linear up to 12 h, followed by a slight decline in the permeability further. The rate of drug permeation through corneal membrane was higher than that in the stomach and rectal membranes. Accordingly, the steady state flux and corresponding permeability coefficient were higher in corneal membrane for both the formulations. This is mainly attributed to the considerable variation in membrane properties like thickness, presence of pores and its diameter and its biological composition.61 Corneal membrane being thin and transparent exhibited higher permeability coefficient and flux compared to stomach and rectal membranes. PERS-2 demonstrated higher permeation and membrane flux than the PEC-3 in situ gel. This may be due to the improved diffusion and penetration of smaller (around 100 nm) hydrophobic nanoparticles in the PERS-2 in situ gel formulation. | ||
| Fig. 10 Ex vivo permeation studies (a) PEC in situ gels (b) PERS in situ gels. | ||
| Formulation code | Membrane | Q24 (μg cm−2) | Jss (μg cm−2 h−1) | Kp (cm h−1 × 10−3) | TL (h) |
|---|---|---|---|---|---|
| PEC-3 | Rectal | 1061.37 ± 144.80 | 54.7715 ± 6.81 | 5.47715 ± 0.68 | 0.69 ± 0.26 |
| Stomach | 1688.50 ± 136.28 | 92.894 ± 8.02 | 9.2894 ± 0.80 | 0.17 ± 0.09 | |
| Cornea | 2451.66 ± 178.45 | 139.805 ± 17.24 | 13.9805 ± 1.72 | 0.18 ± 0.15 | |
| PERS-2 | Rectal | 1315 ± 140.02 | 74.387 ± 8.64 | 7.4387 ± 0.86 | 0.23 ± 0.07 |
| Stomach | 2313 ± 199.23 | 131.53 ± 14.53 | 13.153 ± 1.45 | 0.38 ± 0.17 | |
| Cornea | 3020 ± 122.78 | 177.705 ± 11.10 | 17.7705 ± 1.11 | 0.22 ± 0.005 |
Statistical studies
Statistically significant difference (p < 0.05) in the percentage of drug release with respect to different media at different time points was depicted by two-way ANOVA for the in vitro release data of the formulations.The values of Q, Jss and Kp were significantly different (P < 0.05) for PERS-2 and PEC-3 formulations for all the membranes as predicted for the ex vivo permeation study statistics.
The polymers used in the nanoparticle formulations namely poly vinyl pyrrolidone, ethyl cellulose and eudragit RSPO and the gelling polymer poloxamer are biocompatible, non-toxic and non-irritant, hence approved by FDA and used in food and pharmaceuticals through oral and topical administrations.57,62–64 Eudragit containing ester linkage could be easily degraded by esterase enzyme and poly vinyl pyrrolidone undergoes hydrolytic degradation. Ethyl cellulose is poorly absorbed and not metabolized hence, excreted unchanged through faecal (96%) and urine (<5%).65 PVP and poloxamer has been reported to be excreted mainly by renal clearance mechanism.66,67
Uptake of nanoparticles by HCE cells
Internalization and accumulation of the hybrid polymer blend nanoparticles (AF1 and AF2) in to the HCE cells was spontaneous and dynamic. Cell uptake of nanoparticles was time dependent, since there was increase in the fluorescence nanoparticles inside the cell and into the nucleus with increase in incubation time from 30 min to 1 h and 3 h.68 The results were in accordance with the previous reports where the uptake of the curcumin nanoparticles increased during 10 min to 3 h, whereas the free curcumin was rapidly up taken by cells within 10 min after which the uptake decreased over time.69 It has also been confirmed that curcumin nanoparticles exhibited more efficient uptake and retention in the cells than the free curcumin.70 With 75% entrapment efficiency of curcumin in the formulated hybrid polymer blend nanoparticles, only 5% of free curcumin was released during dialysis in the cell media at the end of 3 h. This could be due to the slow diffusion of hydrophobic molecules and very less hydrolysis of polymer PVP during the initial period. Hence, the green fluorescence inside the cells could be attributed mainly due to the entry of dye loaded nanoparticles along with lesser proportion of the free curcumin. Higher amount of nanoparticles depicted in the cytoplasm (Fig. 11) could favour the system, since it is the major site for drug activation. There was no significant difference between the cellular uptake of PVP–EC and PVP–ERS nanoparticles at the end of 3 h, as both the systems were designed with combination of amphiphilic and hydrophobic polymers which favoured its entry into cells.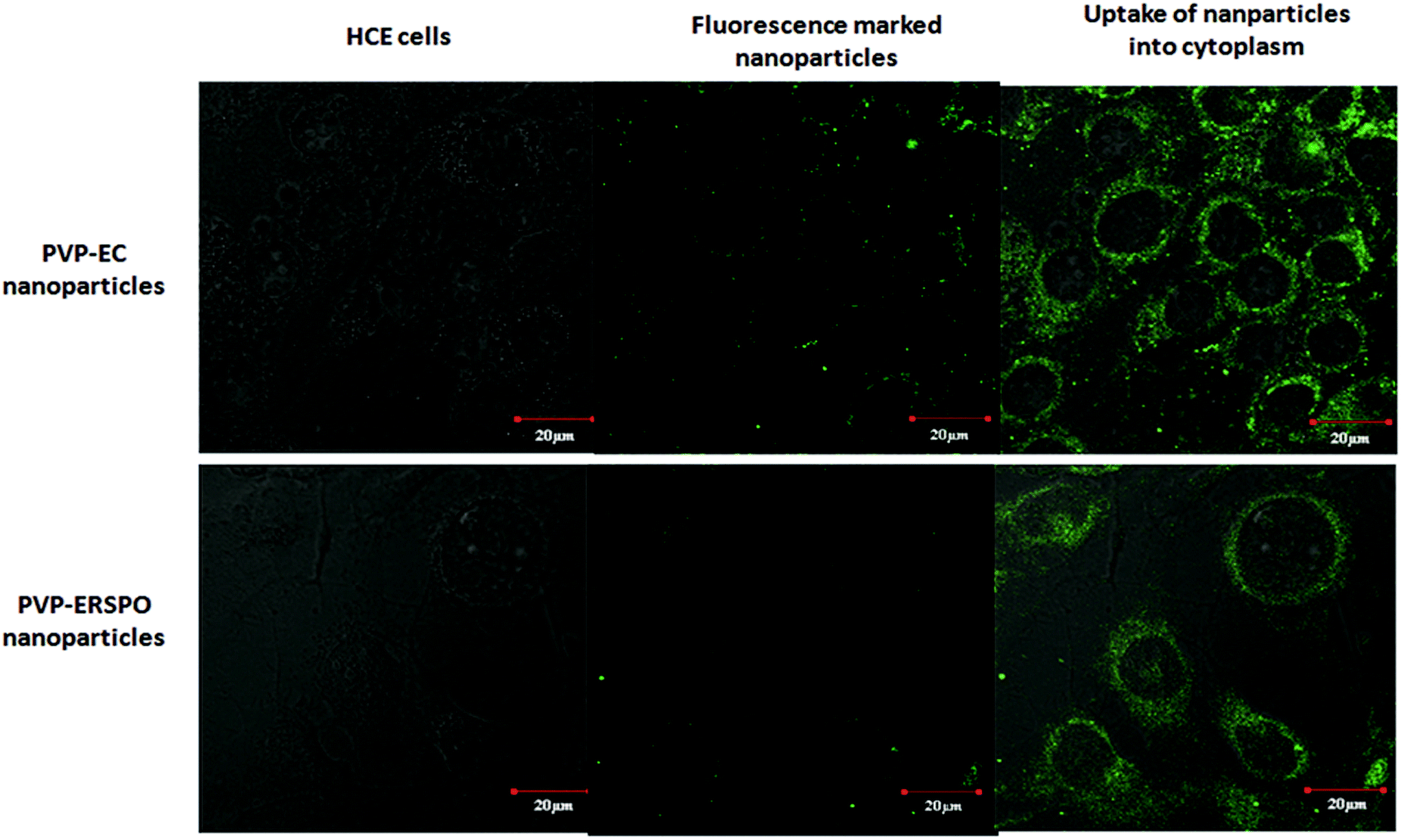 | ||
| Fig. 11 Cellular uptake studies of the hybrid polymer blended nanoparticles. | ||
The cytotoxicity of the developed hybrid polymer blend nanoparticles of Acyclovir has been studied by MTS assay using epithelial cell lines and reported in our earlier work.71 The polymer blend samples and the nanoparticle formulations showed >90% cell viability at concentrations up to 50 μg mL−1. The gelling polymer Pluronic used for incorporation of the nanoparticles has been reported to be non-toxic, non-irritant and non-sensitizing to mouse, rats, dogs and human through various routes. As Pluronic is primarily distributed in extracellular water with little or no uptake by red blood cells, no hemolysis of cells and no symptoms of adverse effects were found during the acute animal toxicity studies.72–74
Characteristics of the foam spray
The foam sprayed out of the canisters was stable and white in colour. Propellant plays vital role in maintaining the foam integrity and uniform dispensing of the foam. The characterization results of the developed foam spray containing Acyclovir loaded polymeric nanoparticles are shown in Table 4.| Formulations tests | PEC-3 | PERS-2 |
|---|---|---|
| Bubble size (μm) | 57.19 ± 17 | 69 ± 28 |
| Relative foam density | 0.071 ± 0.004 | 0.064 ± 0.002 |
| Clarity | Translucent | Translucent |
| Collapse time (min) | 95 ± 4 | 90 ± 5 |
| pH | 6.9 | 7.16 |
| Drug content per puff (mg per 10 mL) | 97.2 ± 12.7 | 101.4 ± 14.06 |
Immediately after dispensing out, the bubble size of the foam was found to be narrow in the range between 55–70 μm. The size of the bubbles increased with respect to time, grown larger and finally ruptured out due to thinning of the film.75 The foam was clear, colourless and translucent in nature. The relative foam density of the PEC-3 and PERS-2 formulations was found to be 0.06 and 0.07, respectively.
The foam was found to be in the grading scale of #1,27 possessing the characteristics of stable, fine bubbles with few coarser bubbles on the surface, then slightly coarser over time. Also the foam appeared to be creamy initially and slowly converted into thin layer, which could favour its spreadability upon contact with the mucous layer through vaginal or rectal application.76 The time taken for the complete collapse of the bubbles was found to be 90–95 minutes (Fig. 12), wherein the formed bubbles were breaking slowly due to the presence of surfactant (Pluronic) in the aqueous phase which gradually decreased the bubble film thickness resulting in rupture of the bubbles.77 The drug content per puff of the foam was found to be in the range of 90–110 mg per 10 mL.
 | ||
| Fig. 12 Appearance and bubble collapse time of foam spray. | ||
Conclusions
The expected therapeutic level of drugs could be predicted with the pharmacokinetic data and thus controlled release systems could be designed to provide pre-determined level of loading and maintenance doses. Such significance could result in remarkable dose reduction, especially for the potent drugs. With this contest, the dosage form of Acyclovir loaded polymeric nanoparticles incorporated into in situ gel was synthesized and optimized for recommended drug entrapment level and controlled release pattern. The obtained data revealed that the optimized formulations meet the required dose release for 20 h to ensure more appropriate once daily administration, as predicted by the pharmacokinetic modeling. It is proved to be a unique dosage for site specific local action in eye, nose, rectal, vaginal, topical and also for the systemic treatment of herpes simplex virus infections. Development of foam spray for vaginal or rectal administration could increase the retention time of drug at applied surface and improve patient compliance. Reduction in the daily dose (conventional oral tablets) to the tune of 10 times is a noteworthy feature of this system, that could decrease the overall side effects, unwanted wastage of drug, drug resistance and reoccurrence of the infections.Acknowledgements
This work was supported by Research and Modernization Project #1 and T.R.R. research scheme, SASTRA University, Thanjavur. We thank Mr Alfred Raymond, Aerosol Division, Sara Industries, Coimbatore, for helping in formulation of foam spray system, and Dr E. Koperundevi, Senior Assistant Professor, Department of English, School of Humanities & Sciences, SASTRA University, for language editing of this manuscript.Notes and references
- L. B. Strick, A. Wald and C. Celum, Clin. Infect. Dis., 2006, 43, 347–356 CrossRef PubMed.
- B. S. Anand, J. M. Hill, S. Dey, K. Maruyama, P. S. Bhattacharjee, M. E. Myles, Y. E. Nashed and A. K. Mitra, Invest. Ophthalmol. Visual Sci., 2003, 44, 2529–2534 Search PubMed.
- K. Suresh, Z. Xiadong, T. S. Ravi and A. K. Mitra, Ophthalmol. Eye Dis., 2010, 2, 43–56 CAS.
- B. Bareiss, M. Ghorbani, F. Li, J. A. Blake, J. C. Scaiano, J. Zhang, C. Deng, K. Merrett, J. L. Harden, F. Diaz-Mitoma and M. Griffith, Open Tissue Eng. Regener. Med. J., 2010, 3, 10–17 CrossRef CAS.
- D. Lembo, S. Swaminathan, M. Donalisio, A. Civra, L. Pastero, D. Aquilano, P. Vavia, F. Trotta and R. Cavalli, Int. J. Pharm., 2013, 443, 262–272 CrossRef CAS PubMed.
- G. S. El-Fekya, G. Zayedb and A. R. H. Farrag, Int. J. Pharm. Pharm. Sci., 2013, 5, 213–219 Search PubMed.
- T. S. Anuradha, K. Prakasam and A. Geethalakshmi, Int. J. Pharmacol. Clin. Sci., 2013, 2, 342–349 Search PubMed.
- B. Chaudhary and S. Verma, Sci. World J., 2014 DOI:10.1155/2014/280928 accessed January 2015.
- A. Gandhi, S. Jana and K. K. Sen, Int. J. Biol. Macromol., 2014, 67, 478–482 CrossRef CAS PubMed.
- S. Shahsavari, E. V. Farahani, M. Ardjmand and F. A. Dorkoosh, Curr. Nanosci., 2014, 10, 521–531 CrossRef CAS.
- Y. S. R. Krishnaiah, X. Xu, Z. Rahman, Y. Yang, U. Katragadda, R. Lionberger, J. R. Peters, K. Uhl and M. A. Khan, Int. J. Pharm., 2014, 475, 110–122 CrossRef CAS PubMed.
- R. J. Whitley, D. W. Kimberlin and B. Roizman, Clin. Infect. Dis., 1998, 26, 541–555 CrossRef CAS.
- J. E. Reardon and T. Spector, Adv. Pharmacol., 1991, 22, 1–27 CAS.
- Acyclovir – Data sheet, http://www.medsafe.govt.nz/profs/datasheet/g/Globalaciclovirtab.htm, accessed September 2014.
- M. Tod, F. Lokiec, R. Bidault, F. Debony, O. Petitjean and Y. Aujard, Antimicrob. Agents Chemother., 2001, 45, 150–157 CrossRef CAS PubMed.
- P. Mehta, Pediatr. Infect. Dis., 2013, 5, 178–180 CrossRef PubMed.
- C. Yildiz, Y. Ozsurekci, S. Gucer, A. B. Cengiz and R. Topaloglu, CEN Case Rep., 2013, 2, 38–40 CrossRef PubMed.
- L. Zeng, C. E. Nath, E. Y. L. Blair, P. J. Shaw, K. Stephen, J. W. Earl, J. C. Coakley and A. J. McLachlan, Antimicrob. Agents Chemother., 2009, 53, 2918–2927 CrossRef CAS PubMed.
- K. Kawakami, Adv. Drug Delivery Rev., 2012, 64, 480–495 CrossRef CAS PubMed.
- M. R. Shaik, M. Korsapati and D. Panati, Int. J. Pharm. Sci., 2012, 2, 112–116 Search PubMed.
- C. Luschmann, J. Tessmar, S. Schoeberl, O. Strauss, C. Framme, K. Luschmann and A. Goepferich, Eur. J. Pharm. Sci., 2013, 50, 385–392 CrossRef CAS PubMed.
- J. Singh, G. Chhabra and K. Pathak, Drug Dev. Ind. Pharm., 2014, 40, 1223–1232 CrossRef CAS PubMed.
- S. Xu, W. Wang, X. Li, J. Liu, A. Dong and L. Deng, Eur. J. Pharm. Sci., 2014, 62, 267–273 CrossRef CAS PubMed.
- Y. Liu, Y. Y. Zhu, G. Wei and W. Y. Lu, Eur. J. Pharm. Sci., 2009, 37, 306–312 CrossRef CAS PubMed.
- Y. Yuan, Y. Cui, L. Zhang, H. Zhu, Y. S. Guo, B. Zhong, X. Hu, L. Zhang, X. Wang and L. Chen, Int. J. Pharm., 2012, 430, 114–119 CrossRef CAS PubMed.
- R. K. Kleppinger, Pa. Med. J., 1965, 68, 31–34 CAS.
- Z. A. Albert and T. H. Barry, US Pat., US7141237 B2, 2006.
- P. Susantakumar, G. Ajay and S. Piush, J. Bioequivalence Bioavailability, 2006, 3, 128–138 Search PubMed.
- S. Basak and K. J. B. Bhusan, Internet J. Pharmacol., 2009, 8, 4391 Search PubMed https://ispub.com/IJPHARM/8/2/4391, accessed January 2015.
- U. V. Bhosale and K. Devi, RGUHS J. Pharm. Sci., 2011, 1, 85–92 Search PubMed.
- K. Selvakumar and A. V. Yadav, Int. J. PharmTech Res., 2009, 1, 179–183 Search PubMed.
- S. Nie, W. L. W. Hsiao, W. Pan and Z. Yang, Int. J. Nanomed., 2011, 6, 151–166 CAS.
- P. K. Kolsure and B. Rajkapoor, Int. J. Curr. Pharm. Res., 2011, 8, 8–14 Search PubMed.
- S. S. Pisal, A. R. Paradkar, K. R. Mahadik and S. S. Kadam, Int. J. Pharm., 2004, 270, 37–45 CrossRef CAS PubMed.
- E. C. Mohan, J. M. Kandukuri and V. Allenki, J. Pharm. Res., 2009, 2, 1089–1094 CAS.
- S. Dey, B. Mazumder and J. R. Patel, Int. J. Pharm. Sci. Drug Res., 2009, 1, 13–18 CAS.
- D. Jasmine and L. Vinod, Int. J. Pharm., 2002, 233, 51–59 CrossRef.
- M. W. Trumbore, R. M. Gurge and J. C. Hirsh, US Pat., 0154402 A1, 2007.
- K. H. Ramteke, S. S. Gunjal and Y. P. Sharma, J. Pharm. Sci. Innovation, 2012, 1, 44–49 Search PubMed.
- B. N. Vedhahari, N. Narayanan and K. Dhevendaran, Chem. Pap., 2015, 69, 358–367 Search PubMed.
- K. Malarvizhi, D. Ramyadevi, R. Alfred and B. N. Vedhahari, Int. J. Sci. Eng. Technol., 2014, 3, 109–115 CAS.
- N. Chandana, H. Gopinath, D. Bhowmik, I. Williamkeri and T. Reddy, J. Chem. Pharm. Sci., 2013, 6, 13–21 Search PubMed.
- R. Katara and D. K. Majumdar, Colloids Surf., B, 2013, 103, 455–462 CrossRef CAS PubMed.
- S. J. Park, G. Choo, S. Hwang and M. Kim, Arch. Pharmacal Res., 2013, 36, 593–601 CrossRef CAS PubMed.
- V. Iole, F. Ilaria, V. R. Maria and B. Andrea, Nanotechnology, 2013, 24, 155503, DOI:10.1088/0957-4484/24/15/155503.
- Y. Haizhou, Q. Xiaoyan, P. N. Suzana and P. Klaus-Viktor, Nat. Commun., 2014, 5, 4110, DOI:10.1038/ncomms5110.
- J. D. Clogston and A. K. Patri, Methods Mol. Biol., 2011, 697, 63–70 CAS.
- Z. P. Liu, L. Cui, D. G. Yu, Z. X. Zhao and L. Chen, Int. J. Nanomed., 2014, 9, 1967–1977 Search PubMed.
- S. Shahi, A. Sonawane, S. Vanamore and N. Zadbuke, J. Appl. Pharm. Sci., 2013, 3, 65–74 Search PubMed.
- D. G. Yu, L. M. Zhu, C. J. Branford-White, J. H. Yang, X. Wang, Y. Li and W. Qian, Int. J. Nanomed., 2011, 6, 3271–3280 CrossRef CAS PubMed.
- D. Ramyadevi and P. Sandhya, Drug Delivery, 2013, 1–17, DOI:10.3109/10717544.2013.839368.
- F. Tirnaksiz and J. R. Robinson, Pharmazie, 2005, 60, 518–523 CAS.
- A. Cabana, A. Ait-Kadi and J. Juhasz, J. Colloid Interface Sci., 1997, 190, 307–312 CrossRef CAS.
- E. J. Riccia, L. O. Lunardib, D. M. A. Nanclaresb and J. M. Marchettia, Int. J. Pharm., 2005, 288, 235–244 CrossRef PubMed.
- J. J. Escobar-Chavez, M. Lopez-Cervantes, A. Naik, Y. N. Kalia, D. Quintanar-Guerrero and A. Ganem-Quintanar, J. Pharm. Pharm. Sci., 2006, 9, 339–358 CAS.
- J. M. Barichello, M. Morishita, K. Takayama and T. Nagai, Int. J. Pharm., 1999, 184, 189–198 CrossRef CAS.
- R. K. Chang, Y. Peng and A. J. Shukla, in Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and S. C. Owen, Pharmaceutical Press, London, 5th edn, 2006, pp. 553–560 Search PubMed.
- D. R. Naik and J. P. Raval, J. Saudi Chem. Soc., 2012 DOI:10.1016/j.jscs.2012.09.020 accessed January 2015, in press.
- D. Suvakanta, N. M. Padala, N. Lilakanta and C. Prasanta, Acta Poloniae Pharmaceutica - Drug Research, 2010, 67, 217–223 Search PubMed.
- T. Ur-Rehman, Ph.D. Thesis, Umea University, Sweden, 2011.
- N. G. Shiow-Fern, J. Rouse, D. Sanderson and G. Eccleston, Pharmaceutics, 2010, 2, 209–223 CrossRef PubMed.
- J. H. Collett, in Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and S. C. Owen, Pharmaceutical Press, London, 5th edn, 2006, pp. 535–538 Search PubMed.
- A. H. Kibbe, in Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and S. C. Owen, Pharmaceutical Press, London, 5th edn, 2006, pp. 611–616 Search PubMed.
- T. C. Dahl, in Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and S. C. Owen, Pharmaceutical Press, London, 5th edn, 2006, pp. 278–282 Search PubMed.
- Modified Celluloses, Compendium amendment 9/FNP 52 add.9/192, 2001 Joint Expert Committee on Food additives of World Health Organization, http://www.inchem.org/documents/jecfa/jecmono/v26je08.htm, accessed Jan 2015.
- Christina L. Burnett, Cosmetic Ingredient Review, 2013 www.cir-safety.org/sites/default/files/PVP_RR.pdf, accessed Jan 2015.
- S. M. Moghimi and A. C. Hunter, Trends Biotechnol., 2000, 18, 412–420 CrossRef CAS.
- P. Anand, H. B. Nair, B. Sung, A. B. Kunnumakkara, V. R. Yadav, R. R. Tekmal and B. B. Aggarwal, Biochem. Pharmacol., 2010, 79, 330–338 CrossRef CAS PubMed.
- S. Jiabei, B. Chao, M. C. Hok, S. Shaoping, Z. Qingwen and Z. Ying, Colloids Surf., B, 2013, 111, 367–375 CrossRef PubMed.
- A. Preeta, B. N. Harish, S. Bokyung, B. K. Ajaikumar, R. Y. Vivek, R. T. Rajeshwar and B. A. Bharat, Biochem. Pharmacol., 2010, 79, 330–338 CrossRef PubMed.
- D. Ramyadevi and K. S. Rajan, J. Taiwan Inst. Chem. Eng., 2015 DOI:10.1016/j.jtice.2014.12.036 in press.
- J. H. Collett, in Handbook of Pharmaceutical Excipients, ed. R. C. Rowe, P. J. Sheskey and S. C. Owen, Pharmaceutical Press, London, 5th edn, 2006, pp. 535–538 Search PubMed.
- J. M. Grindel, T. Jaworski, O. Piraner, R. M. Emanuele and M. Balasubramanian, J. Pharm. Sci., 2002, 91, 1936–1947 CrossRef CAS PubMed.
- S. D. Singh-Joy and V. C. McLain, Int. J. Toxicol., 2008, 27, 93–128 CrossRef PubMed.
- K. F. Popp and E. R. Yuhas, US Pat., US2007/7186416, 2007.
- C. Huang, X. Yang and X. Wang, Chem. Pap., 2006, 60, 102–110 CrossRef CAS PubMed.
- Z. Yanjun, B. B. Marc and A. J. Stuart, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 227–236 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Video file to explain mechanism of gelation of Pluronic system with polymeric nanoparticles is attached. See DOI: 10.1039/c4ra16537c |
| This journal is © The Royal Society of Chemistry 2015 |