
Hybrid polypeptide hydrogels produced via native chemical ligation
Zhiping Fan,
Yemin Zhang,
Jinkai Ji and
Xinsong Li*
School of Chemistry and Chemical Engineering, Southeast University, Nanjing 210018, China. E-mail: lixs@seu.edu.cn
First published on 26th January 2015
Abstract
Biocompatible crosslinking is a key approach for developing biomedical hydrogels and scaffolds. In this report, native chemical ligation (NCL) was utilized to prepare biocompatible and biodegradable hydrogel using naturally derived poly(γ-glutamic acid) and ε-poly-lysine as the backbone without any additive and byproduct. First, thiolactone grafted poly(γ-glutamic acid) (PGA-HC) and cysteine grafted ε-poly-lysine (EPL-C) precursors were synthesized. Their structure was confirmed by nuclear magnetic resonance (NMR). After that, NCL crosslinking of PGA-HC and EPL-C precursors was triggered by simply blending their buffer solutions without any additive at room temperature, resulting in a hybrid polypeptide hydrogel. The crosslinking approach was verified by Fourier transform infrared spectroscopy (FTIR) analysis. The equilibrium water content, morphology, degradation rate and mechanical properties of the hybrid hydrogels were characterized in detail. The results revealed the NCL hybrid hydrogels had tunable gelation time, water content and mechanical properties by adjusting precursor composition. Furthermore, the biocompatibility of hybrid hydrogels was confirmed by MTT assay. These characteristics provide a potential opportunity for the NCL hybrid polypeptide hydrogels as wound dressings, skin fillings, drug delivery vehicles and tissue regeneration matrices.
1 Introduction
Hydrogels are three-dimensional networks capable of absorbing large amounts of water or biological fluids, which have been widely investigated for biomedical applications including drug delivery, cell culture, and tissue engineering biomedical devices.1–15 Recently, because of their biocompatibility and biodegradability, natural biopolymer hydrogels crosslinked under physiological conditions have been intensively studied for regeneration scaffolds and surgical implantations.16,17In the past decades, different methods have been developed to prepare hydrogels. These methods can be classified into two categories: (1) physical crosslinking such as ionic crosslinking18,19 and hydrophobic interactions crosslinking;20–22 (2) chemical crosslinking such as photo-crosslinking,23–25 Michael-type reactions,26,27 Schiff-base reactions28,29 and “click” reactions.28–32 Chemical crosslinking methods are commonly used in the preparation of hydrogels, however the addition of crosslinkers and the generation of residual molecules during crosslinking often reduce the biocompatibility of the hydrogels severely. While, physically crosslinked hydrogels do not have uniform and stable internal structure. Therefore, it remains a challenge to prepare natural biopolymer hydrogels with excellent biocompatibility under mild reaction conditions.
The reaction between a thioester and a N-terminal-Cys, which yields an S-acyl covalent intermediate that spontaneously undergoes an S- to N-acyl migration to form a new amide bond through a five-member ring intermediate, could carry out under physiological conditions.33 This reaction is defined as native chemical ligation (NCL) which has been reported mostly in synthesis of special structural peptides and proteins.34–38 It owns many attractive features including chemoselectivity, high efficiency and mildness reactivity. Moreover, with special structural thioester, two thiol side chains could be released as a consequence of rearrangement in NCL approach,39 which is allowed further modification. However, the utilizing NCL reaction to prepare biomedical hydrogels from natural biopolymers is rarely reported in the literatures.
ε-Poly-lysine (ε-PL, EPL) is a soluble and biodegradable biopolymer, which was found by two Japanese researchers in 1977.40 Although EPL is a commonly known pH sensitive polypeptide, EPL containing hydrogels have received little attention, which is mainly due to the concern on the reported cytotoxicity of free EPL to cells.41,42 However, many recent reports have shown the EPL containing hydrogels exhibited good cytocompatibility,43 good integration with peripheral tissues and in-growth of cells into the hydrogels.44 All these studies indicated that EPL containing hydrogels are promising biomaterials for biomedical applications. Poly(γ-glutamic acid) (PGA) is a natural biopolymer polymerized linearly from glutamic acid, which is known as a main adhesive component of natto. It possesses good solubility, biocompatibility and could degrade into nontoxic short peptides and amino acid monomers. Because of these advantages, the PGA derived materials have been explored for numerous biomedical applications, such as drug release carrier, haemostatic agent and soft tissue augmentation.17,45–47
In this research, a novel hybrid polypeptide hydrogel crosslinked via NCL method was investigated. Two functional precursors were first synthesized, whose structures were subsequently confirmed by 1H NMR. After that, crosslinking of the two precursors was triggered by simply blending their buffer solutions at room temperature resulting in the formation of final hydrogels. In addition, the performances including the equilibrium water content, morphology, degradation rate, swelling ratio and mechanical properties of the NCL hybrid hydrogels were then discussed in detail. Furthermore, in vitro cytotoxicity of the sterilized NCL hybrid hydrogel was evaluated by MTT assay.
2 Materials and methods
2.1 Materials
EPL (MW 4000 Da) was obtained from Bainafo Bioengineering Co., Ltd. (Zhengzhou, China). PGA (MW 1000 kDa) was provided by Shineking Biotechnology Co., Ltd. (Nanjing, China). Homocysteine thiolactone hydrochloride (HC) was purchased from TCI (Shanghai, China). Trifluoroacetic acid (TFA) was obtained from Adamas-beta Chemical Co (Shanghai, China). Triisopropylsilane (TIS), 1-ethyl-(3-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) and protected amino acids were supplied by GL Biochem Ltd. (Shanghai, China).Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), penicillin, L-glutamine, trypsin and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Invitrogen Co. (Carlsbad, CA). L929 mouse fibroblasts cells were obtained from Shanghai Institute of Biochemistry and Cell Biology (SIBCB), Chinese Academy of Science. The L929 cells were cultured in complete growth culture medium prepared with DMEM supplemented with 10% FBS, 1 mM L-glutamine, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a 5% CO2 atmosphere. All the other chemical reagents were supplied by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
2.2 Synthesis of N-terminal cysteine precursor
The synthesis of N-terminal cysteine precursor was performed using protected amino acid Boc-Cys (Trt)-OH (BTC) and EPL as shown in Scheme 1A. BTC (1.81 g, 3.91 mmol) was firstly dissolved in 25 mL dioxan–H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution. EDC (0.75 g, 3.91 mmol) and NHS (0.45 g, 3.91 mmol) were then added to activate the carboxyl of BTC at pH 5 for 2 h. EPL (0.5 g, 3.91 mmol) dissolved in 4 mL dioxan–H2O (1:1, v/v) to form a homogeneous solution, which was subsequently dropped into the activated acid solution. The final reaction mixture was adjusted to pH 6 with 0.1 M NaOH/HCL and stirred overnight. The crude EPL-BTC was precipitated in acetone and then re-dissolved in H2O. The purification cycle of dissolution in H2O and precipitation in acetone was repeated 3 times, followed by drying in vacuum. The resultant product was dissolved in 20 mL TFA–TIS–H2O (95:2.5:2.5, v/v) solution and stirred for 3 h at room temperature.48 Under nitrogen flow, the deprotected precursor (EPL-C) was precipitated in acetone and then re-dissolved in cold water. The cycle of dissolution in H2O and precipitation in acetone was repeated 3 times. After that, the EPL-C precursor dissolved in 20 mL nitrogen bubbled cold water, which was followed frozen at −20 °C and lyophilized to the final EPL-C precursor. 1H NMR was utilized to confirm the structure of EPL-C precursor:49,50 1H NMR (Bruker 500 MHz, D2O) δ 4.15 (s, 1H), 3.18 (d, J = 5.5 Hz, 8H), 2.68–2.40 (d, J = 34.3 Hz, 2H), 1.65 (s, 8H), 1.57–1.43 (m, 8H), 1.33 (t, J = 27.8 Hz, 8H). The degree of substitution (the number of cysteine molecules per 100 repeating units of EPL) was calculated by comparing the ratio of the relative peak integrations of the methylene at 2.68–2.40 ppm of cysteine and two hydrogens of methylene at 3.18 ppm of EPL. The degree of substitution EPL-C precursor is about 24.7%.
:1, v/v) solution. EDC (0.75 g, 3.91 mmol) and NHS (0.45 g, 3.91 mmol) were then added to activate the carboxyl of BTC at pH 5 for 2 h. EPL (0.5 g, 3.91 mmol) dissolved in 4 mL dioxan–H2O (1:1, v/v) to form a homogeneous solution, which was subsequently dropped into the activated acid solution. The final reaction mixture was adjusted to pH 6 with 0.1 M NaOH/HCL and stirred overnight. The crude EPL-BTC was precipitated in acetone and then re-dissolved in H2O. The purification cycle of dissolution in H2O and precipitation in acetone was repeated 3 times, followed by drying in vacuum. The resultant product was dissolved in 20 mL TFA–TIS–H2O (95:2.5:2.5, v/v) solution and stirred for 3 h at room temperature.48 Under nitrogen flow, the deprotected precursor (EPL-C) was precipitated in acetone and then re-dissolved in cold water. The cycle of dissolution in H2O and precipitation in acetone was repeated 3 times. After that, the EPL-C precursor dissolved in 20 mL nitrogen bubbled cold water, which was followed frozen at −20 °C and lyophilized to the final EPL-C precursor. 1H NMR was utilized to confirm the structure of EPL-C precursor:49,50 1H NMR (Bruker 500 MHz, D2O) δ 4.15 (s, 1H), 3.18 (d, J = 5.5 Hz, 8H), 2.68–2.40 (d, J = 34.3 Hz, 2H), 1.65 (s, 8H), 1.57–1.43 (m, 8H), 1.33 (t, J = 27.8 Hz, 8H). The degree of substitution (the number of cysteine molecules per 100 repeating units of EPL) was calculated by comparing the ratio of the relative peak integrations of the methylene at 2.68–2.40 ppm of cysteine and two hydrogens of methylene at 3.18 ppm of EPL. The degree of substitution EPL-C precursor is about 24.7%.
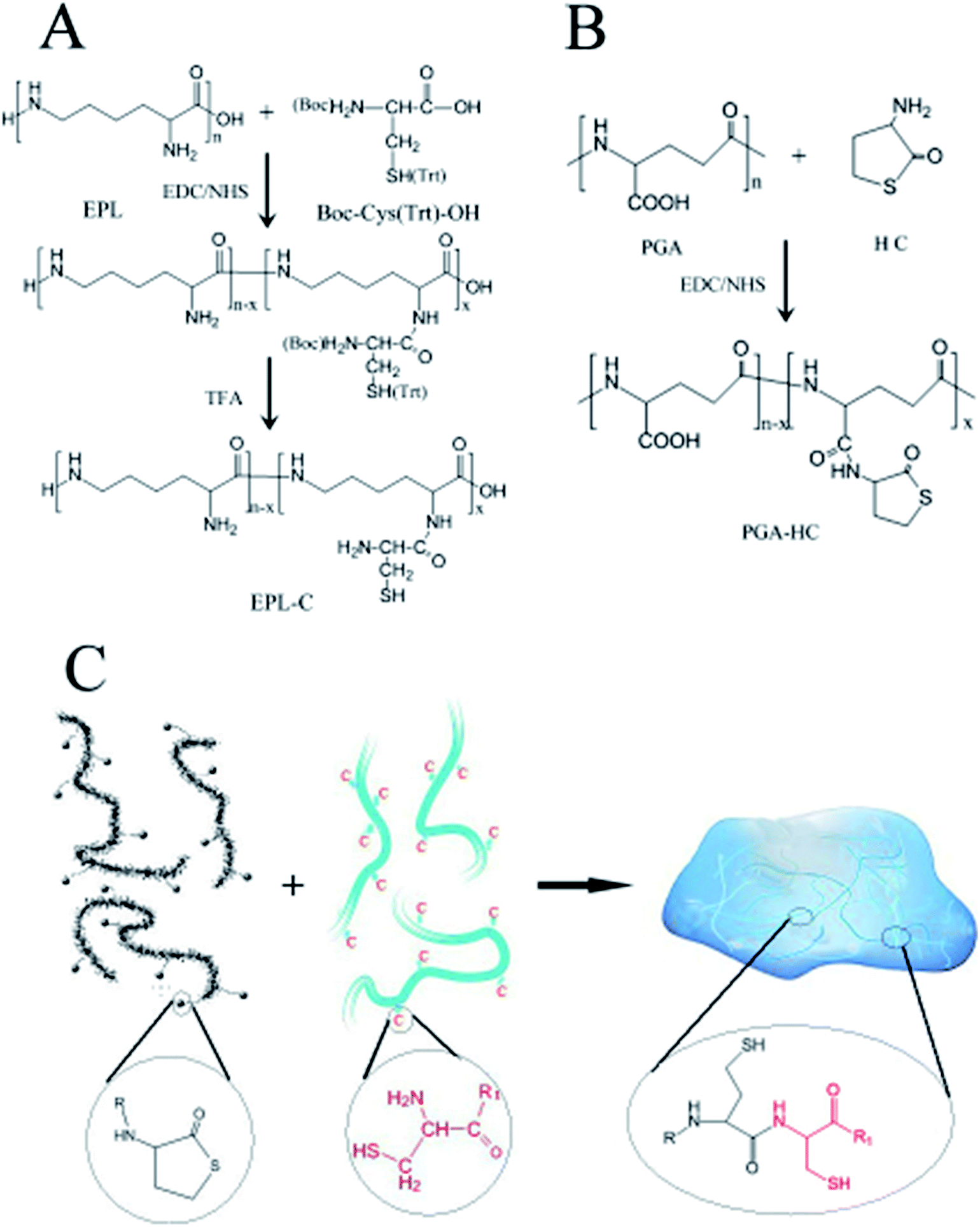 | ||
| Scheme 1 Syntheses of (A) EPL-cysteine precursor; and (B) PGA-thiolactone precursor; (C) the mechanism of NCL hydrogel formation. | ||
2.3 Syntheses of thiolactone precursor
Thiolactone precursor was also synthesized via EDC/NHS chemistry as illustrated in Scheme 1B. Different PGA-HC precursors (P50, P70, P90 and P120) were prepared with different feeding amount of HC (3.875 mmol, 5.425 mmol, 6.975 mmol and 11.16 mmol), and the procedure for the preparation of P90 was followed as an example. Briefly, PGA (1 g, 7.75 mmol) was firstly dissolved in 100 mL distilled water. EDC (4.457 g, 23.25 mmol) and NHS (2.674 g. 23.25 mmol) then added to activate the carboxyl of PGA at pH 5 for 2 h. Homocysteine thiolactone (1.071 g, 6.975 mmol) dissolved in 5 mL H2O to form a homogeneous solution, which was subsequently dropped into the activated acid solution. The reaction mixture was adjusted to pH 6 with 0.1 M NaOH/HCL and stirred overnight at room temperature. Prior to be lyophilized, the solution was dialyzed (MWCO 1000 Da) against distilled water for 1 day. 1H NMR was utilized to confirm the structure of PGA-HC precursor:51,52 1H NMR (Bruker 500 MHz, D2O) δ 4.12 (s, 1H), 3.34 (s, 1H), 2.80 (s, 1H), 2.32 (d, J = 21.9 Hz, 2H), 2.09–1.80 (m, 2H). The degree of substitution (the number of HC molecules per 100 repeating units of PGA) was calculated by comparing the ratio of the relative peak integrations of methylene at 2.80 ppm of HC and the hydrogens of methylene at 1.80–2.09 ppm of PGA. The degree of substitution of P50, P70, P90 and P120 were 27%, 37%, 54% and 42%, respectively.2.4 Hydrogel formation
NCL hydrogels were prepared by simply blending precursors' solutions at room temperature with the composition as shown in Table 1. Briefly, EPL-C (5–10%, w/v) buffer solutions and PGA-HC (5–10%, w/v) aqueous solutions were mixed equal volume at the same concentration. The homogenous solution was following poured into a 5 mm height cylindrical mold which was allowed to form hydrogel at room temperature for 4 h. After released from the mold, the hydrogels were sterilized in 75% ethanol and followed by 3 times of deionized water washing. And then the sterilized hydrogels were used for further characterization. The gelation time was established by the vial tilting method,53 i.e. no flow within 1 min after inverting the vial was regarded as the gel state. No exogenous free thiol or reducing agents were added during the NCL hydrogels formation. The mixtures of P90 and EPL-C before and after crosslinking were used for FTIR analysis. Samples for FTIR spectrum were obtained by making 0.1 g pellets with a ratio of KBr/sample = 99/1. FTIR spectroscopic analysis (Nexus 870, Nicolet) was set on transmission mode with the wavenumber range between 4000 and 350 cm−1.| Hydrogel | EPL-cysteine precursor (w/v) | PGA-thiolactone precursor (w/v) |
|---|---|---|
| 50P-1 | 5% | 5% (P50) |
| 50P-2 | 10% | 10% (P50) |
| 70P-1 | 5% | 5% (P70) |
| 70P-2 | 10% | 10% (P70) |
| 90P-1 | 5% | 5% (P90) |
| 90P-2 | 10% | 10% (P90) |
| 120P-1 | 5% | 5% (P120) |
| 120P-2 | 10% | 10% (P120) |
2.5 Equilibrium water content
The as-prepared hydrogels (50P-1, 50P-2, 70P-1, 70P-2, 90P-1, 90P-2, 120P-1 and 120P-2) were incubated in distilled water at room temperature for 24 h to reach their equilibrium swollen state. After excess surface water was removed, the swollen weight (Ws) of each hydrogel was recorded. Then the samples were lyophilized to complete dryness and the dry weight (Wd) of each samples were weighed. The equilibrium water content (EWC) value was calculated using the following equation:
All the samples were tested in triplicates for each group.
2.6 Scanning electron microscopy
Morphologies of NCL hydrogels were characterized by utilizing scanning electron microscopy (JSM-6360 JEOL, Japan) using 90P-2 as an example. The samples were frozen in a −20 °C freezer and lyophilized in a lyophilizer (Lyovac GT-2, Germany). The lyophilized samples were then positioned on SEM stubs with adhesive tape and sputter coated for 1 min with a thin gold layer. The morphologies of the samples were viewed at 10 kV accelerating voltage.2.7 Mechanical properties
Mechanical properties of the NCL hydrogels (50P-2, 70P-2, 90P-2 and 120P-2) were characterized by the universal testing system (YL-1109, Yuelian Testing Machines Co., Ltd., Dongguan, China) at room temperature. Cylindrical samples with 5 mm height and 10.7 mm diameter prepared in a mold were subjected to compression test with a compression rate of 20 mm min−1. Individual compressive strength was obtained from the load–displacement curve at the break. All the samples were tested in triplicates for each group.2.8 In vitro degradation and swelling
In tests of degradation rate and swelling ratio, 90P-2 hydrogels were immersed in PBS (0.01 M) under constant shaking at 100 rpm for 7, 14, 21 and 28 days at 37 °C to accelerate degradation. The PBS was replaced every week. The hydrogels were washed with distilled water and then lyophilized at each pre-determined time interval. The in vitro degradation rate was calculated by the dry weight after degradation (Wt) divided by the initial dry weight of the gel (W0) as follows:| Fractional mass remaining = (Wt/W0) × 100% |
The swelling ratio (Q) was calculated by the swollen weight after degradation (Ws) divided by the initial weight of the gel (W0) as follows:
| Q = Ws/W0 |
All the samples were tested in triplicates for each group.
2.9 Cytotoxicity evaluation
Evaluation of the cytotoxicity of NCL hydrogels (50P-2, 70P-2, 90P-2 and 120P-2) was accomplished on an extracted solution of the hydrogel via MTT assay according to ISO 10993-5 Standard. Briefly, sterilized hydrogels were extracted using DMEM at an extraction ratio of 1 cm2 mL−1 at 37 °C for 24 h. A 100 μL media suspension containing total of 104 cells and 100 μL extract solution were plated into each well of the 96 well plate then incubated at 37 °C in 5% CO2 atmosphere. On the 1st, 3rd and 5th day, 20 μL of MTT (5 mg mL−1 in PBS) was added for 4 h to allow formation of formazan crystal. After removal of the supernatant, 150 μL DMSO was added to each well and the absorbance was measured at 490 nm using an ELISA reader (Elx800, Bio-Tek Instrument Inc., VT). The results were expressed as percentages relative to the data obtained with blank control. Six samples were tested for each group.3 Results and discussion
3.1 Synthesis of precursors
The synthesis of EPL-C precursor was performed using EDC/NHS chemistry. Comparing the 1H NMR spectra of EPL and EPL-C, the additional signals of the latter at 2.68–2.40 and 4.15 ppm can be respectively attributed to the hydrogens on methylene and methine of cysteine,49 indicating the successful amidation.The EDC/NHS chemistry was also utilized in the synthesis of PGA-HC precursors. Comparing the 1H NMR spectra of PGA and PGA-HC, the additional signals of the latter at 3.34 and 2.80 ppm can be respectively attributed to the hydrogens of two methylenes of thiolactone,51 indicating the successful HC grafting. As reported in the literature,54 the homocysteine thiolactone is prone to ring opening between two molecules in solution, leading to a series of byproducts. So, a set of PGA-HC precursors (P50, P70, P90 and P120) with different HC feeding ratios (50%, 70%, 90% and 120%) were prepared in order to investigate the efficiencies of NCL reactions in this work. Moreover, the thiolactone grafted poly(γ-glutamic acid) could generate two thiol side chains during the NCL crosslinking, which were allowed for further modification with different biofunctional molecules.
3.2 Hydrogels formation
NCL hydrogels were prepared as illustrated in Scheme 1C with composition listed in Table 1. Among all the samples, 90P-2 hydrogels provided a fastest gelation time (≈40 min) in PBS (pH 7.4) at room temperature. The gelation time of other hydrogel samples ranging from 40 to approx. 100 min was also established by the tilting method.53 PGA and EPL are polyanion and polycation, respectively, but the grafted precursors didn't precipitate after mixing at 5% concentration, which is consistent with other reports.55,56 However, at 10% concentration, some white floc appeared at the initial stage of precursor solutions mixed and disappeared completely after the formation of gel. This phenomenon may be attributed to the gradually weakened ions effect during the NCL reactions processing.It's reported that disulfide bond formation and new thioester bond formation by thioester exchange may give rise to hydrogel formation.57 Moreover, the tricarboxyethylphosphine (TCEP) could reduce intermolecular disulfide bonds and 2-mercaptoethanol could exchange the thiol. Both of the agents could lead side reactions formed hydrogel dissolved in water. Therefore, the as-prepared NCL hydrogels were treated with TCEP and 2-mercaptoethanol, respectively. We found that the hydrogels were stable upon addition of an equal volume of 20 mM TCEP in water, and also stable upon addition of 100 mM 2-mercaptoethanol. The results revealed that the side reactions did not affect the hydrogel formation significantly. So, the hydrogel formation was a result of NCL reaction between thiolactone group of PGA-HC and cysteine group of EPL-C. Furthermore, the FTIR spectra of mixtures of P90 and EPL-C before and after crosslinking were also utilized to confirm the occurrence of NCL reaction as shown in Fig. 1. The absorption band at 1662 cm−1 (amide I) increased significantly, indicating the formation of lots of new amide bonds in the NCL hydrogel networks. This result gave another evidence for the mainly crosslinking is the NCL reaction, though we cannot exclude the possibility that disulfide bond formation may contribute partly to the hydrogel formation.
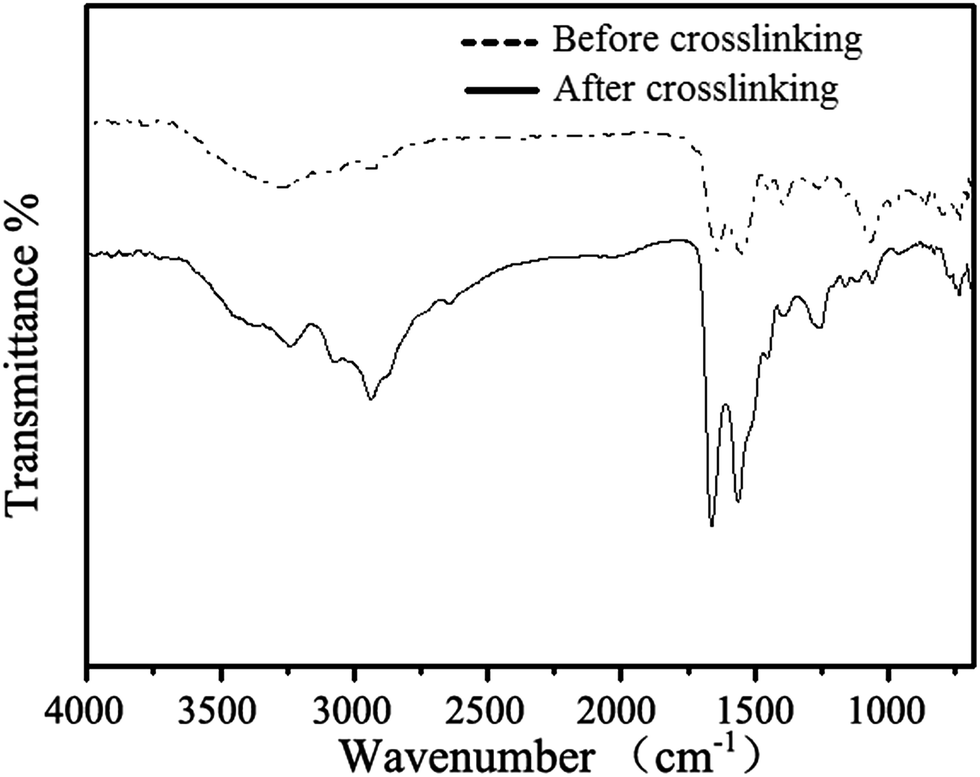 | ||
| Fig. 1 FTIR spectra of mixtures of PGA-HC(P90) and EPL-C before and after crosslinking. | ||
3.3 Equilibrium water content
Fig. 2 illustrated equilibrium water content of different NCL hydrogels. In the first group at 5% concentration, EWC values were decreased with the increasing of precursor's HC grafting ratio. It indicated that the hydrogel network formed more efficiently with the increase of crosslinking density. And the second group at 10% concentration exhibited similar trend with precursor's HC grafting ratio increasing. However, excess feeding ratio of HC may also cause partial destruction of thiolactone in PGA-HC precursor's synthesis.54 It may be the reason that leading a decrease of crosslinking points and resulting in a higher EWC value of 120P hydrogel than that of 90P hydrogel. Among all these samples, 90P-2 hydrogel showed the lowest EWC value of 3.2, which may be ascribed to its highest crosslinking density. The 50P-1 hydrogel was formed at low crosslinking density, resulting in a relatively loose structure with the highest EWC value of 5.6. | ||
| Fig. 2 The equilibrium water contents of different NCL hybrid polypeptide hydrogels. | ||
3.4 Morphology
Pore size and pore interconnectivity are critical parameters determining the performance of hydrogel in tissue engineering. Pore size affects cell attachment, migration, morphology and proliferation; pore structures also have a strong influence on the mechanical properties of matrix, the supply of nutrients and the removal of waste products. In order to characterize the microstructures and morphologies of lyophilized NCL hydrogel, SEM was used to observe their detail. As shown in Fig. 3, the 90P-2 hydrogel used as an example was highly porous with a well-interconnected pore structure. It was characterized by a wide pore size distribution; the estimated pore size was in the range of 10–100 μm. It is assumed that these properties are satisfactory for cell encapsulation and other biomedical applications as reported in the literatures.58,59 | ||
| Fig. 3 SEM images of lyophilized 90P-2 hydrogel samples (left: 100×, right: 1000×). | ||
3.5 Mechanical properties
The compressive strength data of NCL hydrogels was shown in Fig. 4. In 50P-2 group, the structure of hydrogel was relatively unstable due to the lower precursor's HC grafting ratio and crosslinking density, resulting in a lowest compressive strength of 46.7 kPa. The compressive strength of 70P-2 and 90P-2 groups increased gradually with the increasing of precursor's HC grafting ratio. It indicated that the crosslinking density was enhanced, resulting in a more compact hydrogel structure. However, excess feeding ratio of HC may also cause partial destruction of thiolactone in P120 precursor's synthesis,54 which leading to a decrease of crosslinking points and resulting in a lower compressive strength of 120P-2 hydrogel. Among all these groups, 90P-2 showed the highest compressive strength, which is higher than that of 50P-2 by 19.5% (55.8 kPa vs. 46.7 kPa). The 120P-2 group showed a relatively lower compressive strength of 47.7 kPa. This phenomenon was very consistent with those found in the EWC study. | ||
| Fig. 4 The compressive properties of different NCL hybrid polypeptide hydrogels. | ||
3.6 In vitro degradation and swelling
The degradation and swelling properties of the 90P-2 hydrogels were characterized by measuring the hydrogel weight change in PBS at 37 °C, as shown in Fig. 5. It was indicated that the mass of 90P-2 decreased 9% within the first week, and 84% remained after 28 days. While the swelling ratio dropped from 4.18 to 3.85 in the first 7 days and reached to 3.59 after 4 weeks degradation. It revealed the dynamic changes of hydrogel internal structure during the degradation. Both of the curves illustrated the rapid decline in the initial stage, which should be attributed to the relatively unstable network structure in early stage of gel formation. And during the next three weeks, the degradation profile of 90P-2 trended to be more regular and controllable. Besides that, this kind of NCL hydrogels own much longer degradation time and more controllable degradability compared to our previous work.60 One of the most important properties of naturally derived polypeptide is the degradability,17,61 which ideally places this hybrid polypeptide hydrogels as material of choice for drug release and tissue engineering. | ||
| Fig. 5 Degradation and swelling property of 90P-2 hydrogels in PBS at 37 °C; the inset is the photograph of hydrogel after 28 days degradation. | ||
3.7 In vitro cytotoxicity
The in vitro cytotoxicity of NCL hydrogels (50P-2, 70P-2, 90P-2 and 120P-2) was investigated using mouse fibroblast cells (L929) incubated with their extract solutions for 1, 3 and 5 days. The quantitative assessments of their cytotoxicity by MTT assay were shown in Fig. 6. The results revealed that there were no significant differences in cytotoxicity among all the NCL hydrogel groups. It may be attributed to the decreased charge density of the crosslinked networks, which was consistent with the literature.43 Furthermore, the mild crosslinking condition that without any additives and byproducts was also conductive to the enhancement of the biocompatibility of the hydrogels. The calculated value of RGR confirmed the biocompatible nature of NCL hydrogels, which meet the requirements of a potential material for biomedical applications well. | ||
| Fig. 6 The MTT assay of different NCL hybrid polypeptide hydrogels. | ||
4 Conclusion
The naturally derived biopolymers poly(γ-glutamic acid) and ε-poly-lysine have been successfully grafted with thiolactone and cysteine, respectively. Utilizing NCL as the crosslinking strategy, a series of biocompatible hybrid polypeptide hydrogels were prepared under physiological condition without any additive and byproduct, which provided a gelation time ranging from 40 to 100 min at room temperature by altering composition. The hybrid polypeptide hydrogels had adjustable mechanical properties and water content. In addition, the hybrid polypeptide hydrogels showed excellent biocompatibility with controllable degradation. All these characteristics provided a potential opportunity for these hybrid polypeptide hydrogels as numerous biomedical applications, e.g. skin fillings, drug delivery vehicles and tissue regeneration matrix.Acknowledgements
Projects 51373034 and 51073036 were supported by the National Natural Science Foundation of China.Notes and references
- K. Deligkaris, T. S. Tadele, W. Olthuis and A. van den Berg, Sens. Actuators, B, 2010, 147, 765–774 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- A. Sidorenko, T. Krupenkin, A. Taylor, P. Fratzl and J. Aizenberg, Science, 2007, 315, 487–490 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS PubMed.
- C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah and K. S. Anseth, Prog. Polym. Sci., 2008, 33, 167–179 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- X. Jia and K. L. Kiick, Macromol. Biosci., 2009, 9, 140–156 CrossRef CAS PubMed.
- C. He, S. W. Kim and D. S. Lee, J. Controlled Release, 2008, 127, 189–207 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- J. Shi, W. Guobao, H. Chen, W. Zhong, X. Qiu and M. M. Q. Xing, Polym. Chem., 2014, 5, 6180–6189 RSC.
- K. Wang, Q. Fu, X. Chen, Y. Gao and K. Dong, RSC Adv., 2012, 2, 7772–7780 RSC.
- C. Lu, B. Li, N. Liu, G. Wu, H. Gao and J. Ma, RSC Adv., 2014, 4, 50301–50311 RSC.
- M. Boruah, P. Gogoi, A. K. Manhar, M. Khannam, M. Mandal and S. K. Dolui, RSC Adv., 2014, 4, 43865–43873 RSC.
- Z. Fan, Y. Zhang, S. Fang, C. Xu and X. Li, RSC Adv., 2015, 5, 1929–1936 RSC.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef PubMed.
- I. OtaniBajaj and R. Singhal, Bioresour. Technol., 2011, 102, 5551–5561 CrossRef PubMed.
- C. K. Kuo and P. X. Ma, Biomaterials, 2001, 22, 511–521 CrossRef CAS.
- S. R. Van Tomme, M. J. van Steenbergen, S. C. De Smedt, C. F. van Nostrum and W. E. Hennink, Biomaterials, 2005, 26, 2129–2135 CrossRef CAS PubMed.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS.
- B. Jeong, Y. H. Bae and S. W. Kim, J. Controlled Release, 2000, 63, 155–163 CrossRef CAS.
- E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef PubMed.
- S. H. Kim, C. Y. Won and C. C. Chu, J. Biomed. Mater. Res., 1999, 46, 160–170 CrossRef CAS.
- A. K. Burkoth and K. S. Anseth, Biomaterials, 2000, 21, 2395–2404 CrossRef CAS.
- S. H. Kim, C. Y. Won and C. C. Chu, Carbohydr. Polym., 1999, 40, 183–190 CrossRef CAS.
- S. Cai, Y. Liu, X. Zheng Shu and G. D. Prestwich, Biomaterials, 2005, 26, 6054–6067 CrossRef CAS PubMed.
- X. Zheng Shu, Y. Liu, F. S. Palumbo, Y. Luo and G. D. Prestwich, Biomaterials, 2004, 25, 1339–1348 CrossRef PubMed.
- K. Y. Lee, E. Alsberg and D. J. Mooney, J. Biomed. Mater. Res., 2001, 56, 228–233 CrossRef CAS.
- B. Balakrishnan and A. Jayakrishnan, Biomaterials, 2005, 26, 3941–3951 CrossRef PubMed.
- A. Takahashi, Y. Suzuki, T. Suhara, K. Omichi, A. Shimizu, K. Hasegawa, N. Kokudo, S. Ohta and T. Ito, Biomacromolecules, 2013, 14, 3581–3588 CrossRef CAS PubMed.
- G. Huerta-Angeles, M. Němcová, E. Příkopová, D. Šmejkalová, M. Pravda, L. Kučera and V. Velebný, Carbohydr. Polym., 2012, 90, 1704–1711 CrossRef CAS PubMed.
- G. Huerta-Angeles, D. Šmejkalová, D. Chládková, T. Ehlová, R. Buffa and V. Velebný, Carbohydr. Polym., 2011, 84, 1293–1300 CrossRef CAS PubMed.
- T. Wieland, E. Bokelmann, L. Bauer, H. U. Lang and H. Lau, Justus Liebigs Ann. Chem., 1953, 583, 129–149 CrossRef CAS.
- S. E. Paramonov, V. Gauba and J. D. Hartgerink, Macromolecules, 2005, 38, 7555–7561 CrossRef CAS.
- D. Macmillan, Angew. Chem., Int. Ed., 2006, 45, 7668–7672 CrossRef CAS PubMed.
- P. E. Dawson and S. B. Kent, Annu. Rev. Biochem., 2000, 69, 923–960 CrossRef CAS PubMed.
- P. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CAS.
- J. P. Tam, Q. Yu and Z. Miao, Pept. Sci., 1999, 51, 311–332 CrossRef CAS.
- P. B. Messersmith, US Pat. 20110262492 A1, 2011.
- S. Shima and H. Sakai, Agric. Biol. Chem., 1977, 1807–1809 CrossRef CAS.
- S. Choksakulnimitr, S. Masuda, H. Tokuda, Y. Takakura and M. Hashida, J. Controlled Release, 1995, 34, 233–241 CrossRef CAS.
- D. Morgan, V. L. Larvin and J. D. Pearson, J. Cell Sci., 1989, 94, 553–559 CAS.
- L. M. Pakstis, B. Ozbas, K. D. Hales, A. P. Nowak, T. J. Deming and D. Pochan, Biomacromolecules, 2003, 5, 312–318 CrossRef PubMed.
- C.-Y. Yang, B. Song, Y. Ao, A. P. Nowak, R. B. Abelowitz, R. A. Korsak, L. A. Havton, T. J. Deming and M. V. Sofroniew, Biomaterials, 2009, 30, 2881–2898 CrossRef CAS PubMed.
- K. Hoste, E. Schacht and L. Seymour, J. Controlled Release, 2000, 64, 53–61 CrossRef CAS.
- J. M. Buescher and A. Margaritis, Crit. Rev. Biotechnol., 2007, 27, 1–19 CrossRef CAS PubMed.
- C. T. Tsao, C. H. Chang, Y. Y. Lin, M. F. Wu, J. L. Wang, J. L. Han and K. H. Hsieh, Carbohydr. Res., 2010, 345, 1774–1780 CrossRef CAS PubMed.
- J. Su, B.-H. Hu, W. L. Lowe Jr, D. B. Kaufman and P. B. Messersmith, Biomaterials, 2010, 31, 308–314 CrossRef CAS PubMed.
- M. C. de Koning, L. Petersen, J. J. Weterings, M. Overhand, G. A. van der Marel and D. V. Filippov, Tetrahedron, 2006, 62, 3248–3258 CrossRef CAS PubMed.
- C. Zhou, P. Li, X. Qi, A. R. Sharif, Y. F. Poon, Y. Cao, M. W. Chang, S. S. Leong and M. B. Chan-Park, Biomaterials, 2011, 32, 2704–2712 CrossRef CAS PubMed.
- T. W. Steele and W. T. Shier, J. Pharm. Res., 2010, 27, 683–698 CrossRef CAS PubMed.
- G. A. Birrer, A.-M. Cromwick and R. A. Gross, Int. J. Biol. Macromol., 1994, 16, 265–275 CrossRef CAS.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- V. du Vigneaud, W. I. Patterson and M. Hunt, J. Biol. Chem., 1938, 126, 217–231 CAS.
- H. J. Choi, R. Yang and M. Kunioka, J. Appl. Polym. Sci., 1995, 58, 807–814 CrossRef CAS.
- M. Kunioka and H. Choi, J. Environ. Polym. Degrad., 1996, 4, 123–129 CrossRef CAS.
- B.-H. Hu, J. Su and P. B. Messersmith, Biomacromolecules, 2009, 10, 2194–2200 CrossRef CAS PubMed.
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Biomaterials, 2009, 30, 2499–2506 CrossRef CAS PubMed.
- A. Al-Abboodi, J. Fu, P. M. Doran, T. T. Tan and P. P. Chan, Adv. Healthcare Mater., 2014, 3, 725–736 CrossRef CAS PubMed.
- C. Wen, L. Lu and X. Li, Macromol. Mater. Eng., 2014, 299, 504–513 CrossRef CAS.
- E. Pişkin, Biodegradable Polymers in Medicine, Springer, Netherlands, 2002 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |