
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of novel tetra-hetero[8]circulenes: a theoretical study of electronic structure and charge transport characteristics†
Vu Thi
Thu Huong
,
Truong Ba
Tai
* and
Minh Tho
Nguyen
*
Department of Chemistry, University of Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: minh.nguyen@chem.kuleuven.be; truong.batai@chem.kuleuven.be
First published on 24th February 2015
Abstract
In the present work, a series of new tetra-hetero[8]circulenes were theoretically designed in which heteroatoms include O, S, Se and N. Their electronic structure and characteristics of charge transport were investigated using DFT based computational methods. Except for the compounds containing Se-atoms (3a and 3b), all remaining compounds exhibit planar and highly symmetrical structures featuring novel aromatic features: the inner eight-membered ring is anti-aromatic and the outer fused rings are aromatic. The predicted UV spectrum and reduction potential of 1a agree well with available experimental values. Following replacement of H-atoms by F-atoms, the energy levels of the frontier orbitals of 1b–4b are consistently decreased as compared to those of 1a–4a. Based on the calculated properties of electrochemistry and charge transport, the molecules 1a–4a and 4b are suggested to be good candidates for p-type semiconductors. More importantly, the molecules 1b–3b are revealed to be potential ambipolar organic semiconductors.
1. Introduction
[n]Circulenes, which consist of compounds having two concentric annulene rings, have attracted much attention as an intriguing family of aromatic polycyclic hydrocarbons. Three members of this family, including [5]circulene (corannulene),1 [6]circulene (coronene)2 and [7]circulene3 (pleiadannulene), have experimentally been prepared for a long time and the characteristics of their electronic structure, chemical bonding and adsorption spectra were well determined. Although earlier theoretical studies suggested that [8]circulene is unstable,4 this molecule was recently successfully synthesized by Wu and coworkers.5 Their shapes interestingly change from a bowled shape for [5]circulene to a planar structure for [6]circulene and saddled shapes for [7] and [8]circulenes.Recently, the family of hetero[8]circulenes appears to be even more intriguing. Replacing the outside benzene fused-rings by heterocyclic rings such as furan, thiophene… results in planar heteropolycyclic compounds with highly π-conjugated electronic structures which can eventually be used in many important applications. Octathio[8]circulene, named as sunflower, (C16S8) in which all outside fused benzene rings are replaced by eight fused thiophene rings, was for the first time synthesized in 2006,6 and found as a good p-type organic semiconductor by both experiment and theory.7,8 Subsequently, the electronic structure and charge transport properties of its analogues molecules such as C16S4Se4 and C16X8 with X = O, Se, NH, CH2, PH and PF were also extensively studied.9–12 They were suggested as either p-type or n-type organic semiconducting materials with high rate of charge hopping. More recently, Datta and his co-workers13 designed similar molecules, named as Si- and Ge-sulflower, in which C-atoms of C16S8 were replaced by either Si-atoms or Ge-atoms. These molecules were found to be bowled structures and exhibit a large number of metallic-like states with weak optical absorption. In a recent study, we theoretically designed a series of new hetero[8]circulenes containing borole units.14 These heteropolycyclics feature some intriguing aromatic characteristics at both inner and outer rings. Such interesting results stimulated us to further design of new π-conjugated materials based on the [8]circulene framework.
Tetra-hetero[8]circulenes in which only four fused benzene rings are replaced by four fused hetero-rings, have recently emerged as promising π-conjugated materials for organic electronic devices. In fact, tetraoxo[8]circulene was already found by experiment since 1960s,15 but its applicable properties were not properly explored. Recently, Minaev and his co-workers performed a series of theoretical and experimental studies on electronic structure, spectroscopic properties, aromaticity and bonding characteristics of tetraoxa[8] circulenes and its derivatives.16,17 Pittelkow et al.18 reported the preparation of a series of π-extended tetraoxa[8[circulenes and their optical and electrochemical properties. These compounds showed high thermodynamic stability and can be used as promising materials for the development of blue organic light-emitting diodes (OLEDs). More recently, some planar cyclooctatetraenes such as azatrioxa[8]circulenes and diazadioxa[8]circulenes in which one and two O-atoms were replaced by N-atoms were successfully synthesized. Their characteristics of single-crystal X-ray structure, aromaticity, and spectroscopy were investigated in both experiment and theory.19 In other way, Minaev et al.20,21 reported theoretical results on the one- and two-dimensional nano-materials containing tetraoxa[8]circulene monomers. The authors found that these materials can be used as ambipolar organic semiconductors with high mobility for both hole and electron charge carriers.
In order to further explore this intriguing class of heteropolycyclic compounds, we herein performed theoretical studies of structural characteristics, electronic and charge transport properties of tetraoxo[8]circulene 1a and its derivatives in two different series 2a–4a and 1b–4b using DFT-based computational methods. The derivatives are designed by simply replacing oxygen atoms of 1a with sulfur (2a), selenium (3a) and NH-groups (4a). Additionally, replacement of H-atoms of compounds 1a–4a by fluorine results in perfluoronated compounds 1b–4b that are expected to have reduced LUMO energy levels and can thus be used as effective n-type organic semiconductors. Our results show that most of designed molecules exhibit the planar structures with high degree of π-conjugation and high rate of charge hopping.
2. Computational methods
2.1 Electronic structure calculations
Optimizations of the relevant geometries and calculations of their harmonic vibrational frequencies are fully performed using DFT. The hybrid functional B3LYP22–24 are used in conjunction with the 6-31G(d,p) basis sets.25,26 For open-shell systems such as cations and anions, the unrestricted formalism (UB3LYP) is used. This popular functional was proved to be reliable in predicting geometrical parameters and charge transport properties of many π-conjugated systems. For each species considered, the neutral, anionic and cationic states are characterized. All electronic structure calculations are carried out using the Gaussian 09 (ref. 27) suite of programs.2.2 Charge transport properties
In order to probe the charge transport properties, the hopping model is used.28–33 Accordingly, the hole and electron carriers can be jumped between the adjacent molecules of the organic crystals. The rate of charge hopping (K) is estimated by using the Marcus–Hush eqn (1):34 | (1) |
The transfer integral is defined by the following expression (2):35–37
| t = 〈ϕ0,site1i|F|ϕ0,site2j〉 | (2) |
The transfer integral for hole (th) is calculated as half of the energy difference between the HOMO and HOMO − 1 of the dimer, while the transfer integral for electron (te) is half of the energy difference between the LUMO and LUMO + 1 of the dimer. These orbital energy levels are obtained using density functional theory (DFT) with the PW91PW91 functional38,39 in conjunction with the 6-31G(d,p) basis set. This approach has been shown to be suitable and effective in calculations of the transfer integrals of many organic compounds.40–48 We should note that the transfer integral is functional dependent property and their values systemically depend on the fraction of nonlocal HF exchange incorporated in a functional. Sutton et al.48b showed that the amount of HF exchange substantially affects the transfer integral in a linear fashion with respect to the fraction of nonlocal HF exchange incorporated in a standard hybrid functional.
2.3 Reduction and oxidization potentials in solution
The reduction and oxidization potentials of compounds in solution are calculated by using a protocol recently developed by Davis and Fry.49 Firstly, optimizations of geometries and calculations of vibrational frequencies of each compound in the neutral, anionic and cationic charge states are performed at the B3LYP/6-31+G(d) level. Their single point electronic energies are obtained using a larger basis set, B3LYP/6-311++G(2df,2p). Addition of a set of diffuse functions in the basis set is necessary to describe the structures of the ions involved in the evaluation. The SMD/IEF-PCM solvation model50 in acetonitrile solvent is used to probe the solvent effects. These methods were proven to be effective to obtain the reduction and oxidation potentials of PHA compounds in solution.49,51 Davis and Fry49 showed that using eqn (4) and (5) results in a mean absolute deviation (MAD) for the polycyclic aromatic hydrocarbons (PAHs) of 0.023 and 0.032 mV, respectively.The absolute potentials at 298 K are obtained using the following eqn (3):
| EAbs298 = Ecalc298 − 0.03766 | (3) |
The reduction potential is predicted in MeCN solution for the ferrocene–ferrocenium (Fc/Fc+) pair, and the standard redox couple are calculated by using the expressions (4) and (5):
| Reduction: E1/2298 = 1.056EAbs298 − 4.90 | (4) |
| Oxidation: E1/2298 = 0.932EAbs298 − 3.94 | (5) |
3. Results and discussion
3.1 Electronic structure and aromaticity of compounds
Geometric optimizations and calculations of harmonic vibrational frequencies of all compounds are performed using the hybrid functional B3LYP in conjugation with the polarized 6-31G(d,p) basic set. This method was demonstrated to provide reliable geometries and charge transport properties of the organic semiconductors in many previous studies.53–58 The chemical structures of all compounds are shown in Fig. 1, whereas their shapes are given in the ESI file.†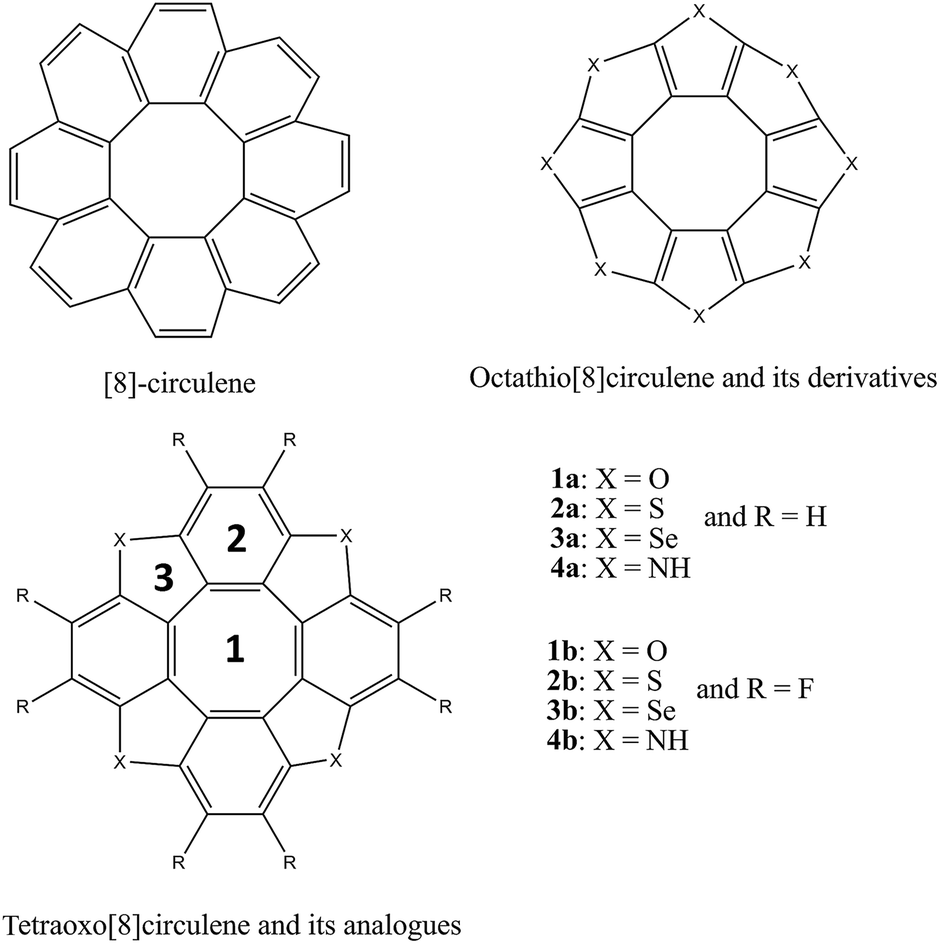 | ||
| Fig. 1 Chemical structure of the [8]circulene and hetero[8]circulenes considered. | ||
Among the eight compounds considered, 1a is the only one which has been well described by both experiment and theory. The computed C–O bond length of 1a of 1.385 Å agrees well with the experimental value of 1.392–1.402 Å.13 The distances between C and hetero atoms of five-membered rings change somewhat, and in the ordering of C–O ∼ C–N < C–S < C–Se. Consequently, our calculations show that the compounds containing furan (1a and 1b), thiophene (2a and 2b) and pyrrole (4a and 4b) exhibit planar structures whereas the compound containing selenophene (3a and 3b) is slightly distorted out of the structural plane due to the large C–Se distances. Following attachment of one excess electron into the molecules to form anionic species (or detaching one electron from the molecules to form cationic species), the geometries of the charged species only change somewhat, except for the case of 3a whose cationic state shows a large distorted shape. Consequently, we can expect that these compounds will have low reorganization energies that can dominate the rate of charge hopping.
Aromaticity is used to describe electron delocalization in compounds. Aromatic compounds usually exhibit high stability, planar and symmetrical structure and good electron delocalization. These are key properties for high charge mobility of π-conjugated organic molecules. Thus, evaluation of aromaticity of a compound gives us more insight into its electronic properties. Aromatic character is evaluated here using the nuclear independent chemical shift (NICS) index.59,60 The NICS(0) values are calculated on ghost atoms located at center of rings 1, 2 and 3 as shown in Fig. 1. It is also known that the NICS(0) values can be affected by distribution of σ-electrons of compounds.54 Thus, the NICS(1) values that are calculated on ghost atoms located at 1 Å distance out of the structural plane can give us more reliable indications.
Computed results in Table 1 showed that the inside eight-membered rings of compounds are anti-aromatic with positive NICS values, whereas all outside rings exhibit aromaticity with negative NICS values. There is negligible difference in aromatic character of molecules with different hetero atoms. In addition, replacing H-atoms of 1a–4a by F-atoms of 1b–4a only change somewhat the aromatic feature of compounds. These aromatic features are similar to those of the sulflower C18S8 and its derivatives.45 Thus it is expected that these compounds are characterized by high π-conjugation, and thereby good ability of intramolecular charge transport.
| Compounds | NICS(0) | NICS(1) | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | |
| 1a | 8.4 | −10.4 | −9.2 | 5.3 | −10.1 | −9.2 |
| 1b | 7.1 | −13.5 | −12.3 | 4.5 | −10.7 | −8.7 |
| 2a | 6.7 | −9.5 | −10.4 | 3.9 | −9.9 | −8.0 |
| 2b | 6.8 | −12.2 | −10.4 | 4.2 | −10.2 | −7.5 |
| 3a | 4.1 | −10.4 | −15.2 | 7.1 | −8.5 | −13.8 |
| 3b | 4.3 | −10.0 | −8.1 | 7.0 | −11.7 | −9.1 |
| 4a | 8.0 | −11.0 | −12.0 | 4.1 | −9.6 | −11.1 |
| 4b | 6.7 | −14.3 | −12.5 | 4.9 | −9.9 | −10.7 |
3.2 Absorption UV spectra of compounds
The absorption UV spectra of all compounds are predicted using the time-dependent (TD-DFT) method with the B3LYP functional in conjunction with the 6-31+G(d,p) basis set. This method was evaluated to give reliable predictions for optical spectra of organic compounds.61,62 The plots of absorption UV spectra of all compounds are displayed in Fig. 2. | ||
| Fig. 2 Plots of the UV spectra of all molecules 1a–4a and 1b–4b. | ||
There is a good agreement between our theoretical predictions and the available experimental and theoretical results. Accordingly, the UV spectrum of 1a shows two local peaks with high intensity centered at 255 and 261 nm which agree well to the experimental value of 260 nm.18 Another local peak is located at 354 nm that is close to the experimental peaks at 358 and 376 nm.18
Our calculations also reveal a local peak at 416 nm which agrees well with the experimental value of 415 nm. This peak actually corresponds to a π–π* transition from HOMO to LUMO. However, this absorption has low intensity in both experimental and theoretical spectra. The low absorption intensity corresponds to a small transition moment between the ground and excited states. The transition moment is derived mainly from the integrals of the dipole moment acting on both states. In this case, it is characterized by the small overlap between the transition orbitals HOMO and LUMO. Similar observations were also found for the UV spectra of remaining compounds.
More interestingly, Fig. 2 shows that the UV spectra of compounds considered tend to be red-shifted when O-atoms of 1a and 1b are replaced by S- and Se-atoms. Two high intensity peaks at 267 and 383 nm were found in the UV spectrum of 2a, whereas the 3a is characterized by two local peaks at 285 and 415 nm. Following perfluorination of compounds 1a–3a, the first local peaks in UV spectra of 1b–3b is red-shifted, while their second local peaks are blue-shifted as compared to those of 1a–3a, respectively. The UV spectra of 4a and 4b are similar to those of 2a and 2b, respectively.
3.3 Reduction and oxidation potentials of compounds in the solution
The values of reduction and oxidation potentials, that are related to the energies of frontier molecular orbitals (FMO), play important role for applicability of organic semiconductors. The energy levels of HOMO (characteristic for p-type semiconductor) and LUMO (for n-type semiconductor) should be close to work functions (WF) in vacuum of electrodes in order to improve their charge transport ability. In experimental studies, some electrodes such as gold (Au) (WF ∼ 5.10 eV), calcium (WF ∼ 2.90 eV), and magnesium (WF ∼ 3.68 eV) are commonly used.63 While the gold electrode is one of the best electrodes for organic electronic device, the calcium and magnesium electrodes are the preferred ones for n-type organic semiconductors due to the fact that this type of materials usually possesses a LUMO energy level of ∼3.0–4.0 eV. In this study, the reduction and oxidation potentials and energy levels of frontier orbitals of the molecules examined are calculated using the SMD/IEF-PCM solvation model64 in acetonitrile solvent (see above). All computed values are given in the Table 2, while the shapes of frontier orbitals are displayed in Fig. 3.| Comp. | E red1/2 | E oxd1/2 | HOMO | LUMO | Gap | Comp. | E red1/2 | E oxd1/2 | HOMO | LUMO | Gap (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | −2.09 | 1.47 | −6.27 | −2.71 | 3.55 | 1b | −1.83 | 1.50 | −6.30 | −2.97 | 3.34 |
| 2a | −2.20 | 1.12 | −5.92 | −2.60 | 3.32 | 2b | −1.82 | 1.44 | −6.24 | −2.98 | 3.26 |
| 3a | −2.11 | 0.87 | −5.67 | −2.69 | 2.98 | 3b | −1.82 | 1.25 | −6.05 | −2.98 | 3.07 |
| 4a | −2.58 | 0.47 | −5.27 | −2.22 | 3.05 | 4b | −2.32 | 0.90 | −5.70 | −2.48 | 3.22 |
 | ||
| Fig. 3 Shapes of HOMOs and LUMOs of molecules and their energy levels in unit of eV. | ||
The reduction potential of 1a is equal to −2.09 eV that is slightly higher than the highest experimental value of −2.39 eV of 1a.18 It can be observed that the compounds containing group VIA elements 1a–3a and 1b–3b have comparable energy levels of frontier orbitals. The LUMO and HOMO of 4a and 4b containing pyrrole units are higher. Additionally, the HOMO energy levels of the 1a–4a vary in a range of (−5.27)–(−6.27) eV, and are in the sequence of 1a < 2a < 3a < 4a. A similar trend is found for the series 1b–4b. These values are close to the work function of 5.1 eV of gold electrode, and consequently they are expected to be good candidates for p-type semiconductors.
Replacement of H-atoms in 1a–4a by F-atoms consistently decreases the energy levels of both HOMO and LUMO of 1b–4b. More importantly, the LUMOs of 1b–3b have similar energy levels and are equal to ∼3.00 eV. These values are very close to that of 2.90 eV of the calcium electrode. The LUMO energy levels of 1a–3a are somewhat higher and close to 2.70 eV. Thus, the perfluorinated compounds 1b–3b can act as ambipolar organic semiconductors. 1a–3a and compounds containing pyrrole 4a and 4b have high LUMOs, and accordingly they can only be suitable for p-type organic semiconducting materials.
3.4 Charge transport properties of compounds
| λh = λ0 + λ+ = [E(M) − E0(M)] + [E(M+) − E0(M+)] | (6) |
| λe = λ0 + λ− = [E(M) − E0(M)] + [E(M−) − E0(M−)] | (7) |
In the present work, the reorganization energies for electron and hole of all compounds are calculated using the B3LYP/6-31G(d,p) method and the computed results are listed in Table 3.
| Comp. | D dimer (Å) | Interaction energy (eV) | Reorganization energy (eV) | Integral transfer (t, meV) | Rate of charge hopping (K, s−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| B3LYP-3D | M06 | Hole | Electron | Hole | Electron | Hole | Electron | ||
| a These values obtained from ref. 10. | |||||||||
| 1a | 3.74 | 1.00 | 1.12 | 0.19 | 0.19 | 36 | 27 | 7.88 × 1012 | 4.50 × 1012 |
| 2a | 3.85 | 1.30 | 1.40 | 0.13 | 0.24 | 7 | 12 | 5.77 × 1011 | 5.27 × 1011 |
| 3a | 3.95 | 1.30 | 1.34 | 0.35 | 0.28 | 22 | 61 | 4.64 × 1011 | 7.47 × 1012 |
| 4a | 3.84 | 1.04 | 1.12 | 0.17 | 0.19 | 7 | 70 | 3.39 × 1011 | 2.78 × 1013 |
| 1b | 3.70 | 1.22 | 1.40 | 0.26 | 0.29 | 25 | 34 | 1.75 × 1012 | 2.16 × 1012 |
| 2b | 3.62 | 1.51 | 1.68 | 0.17 | 0.28 | 28 | 16 | 5.73 × 1012 | 5.79 × 1011 |
| 3b | 3.68 | 1.57 | 1.60 | 0.24 | 0.34 | 49 | 17 | 7.80 × 1012 | 2.93 × 1011 |
| 4b | 3.80 | 1.36 | 1.51 | 0.25 | 0.29 | 7 | 25 | 1.36 × 1011 | 1.21 × 1012 |
| C16S8 a | 3.87 | — | 0.98 | 29 | 6 | ||||
Except for the relatively large λh value of 3a (λh = 0.35 eV, Table 3) which is due to a large distortion of its cationic state, the reorganization energies for hole and for electron of 1a–4a are quite low and vary in a range of 0.17–0.19 eV and 0.19–0.28 eV, respectively. As compared to the computed values of C16(PF)8 (λh = 0.30 eV and λe = 0.22 eV) and perfluoropentacene (PF-PEN) (λh = 0.23 eV and λe = 0.23 eV) which is a typical n-type organic semiconductor, the reorganization energy for hole (λh) of 1a–4a are properly lower. Their λe values are close to those of PF-PEN and C16(PF)8. Although the perfluorinated compounds 1b–4b showed the λh and λe values a bit higher than those of 1a–4a, they are also comparable to those of compounds previously reported.
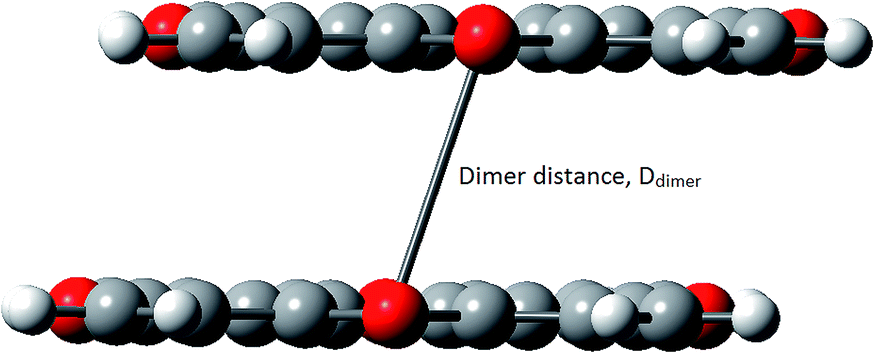 | ||
| Fig. 4 The translational parallel motif of dimers. | ||
The intermolecular interaction energy of dimers is defined by expression (8):
| Eint = 2Emonomer − Edimer | (8) |
Our calculated results are given in Table 3 and compared with calculated properties of the previously reported C16S8. We should note that the molecular structures of the designed compounds 1a–4a and 1b–4b are in fact similar to that of sulflower C16S8. Thus, a comparison between C16S8 and the molecules designed in this work appears useful to understand their charge transport characteristics.
The strong interactions between monomers in dimers of all compounds were observed. The distances between monomers (Ddimer) vary in a small range of 3.62 ÷ 3.95 Å and are close to the value of 3.87 Å of C16S8 in the same interaction motif. The interaction energies between two monomers amount to 1.04–1.57 eV that is properly larger than the value of 0.98 eV of C16S8. Consequently, it can be seen that integral transfers for hole of compounds are comparable to that of C16S8, whereas their integral transfers of electron becomes much larger. Since the C16S8 was reported as good p-type organic semiconductor, the compounds considered in this work are expected to be appropriate for both n-type and p-type semiconductors.
4. Conclusions
We theoretically designed a series of new tetra-hetero[8]circulenes based on an inner eight-membered ring. The electronic structure and characteristics of charge transport of all compounds were investigated using DFT based computational methods. Except for the compounds containing Se-atoms (3a and 3b), all remaining compounds exhibit planar and highly symmetrical structures with a novel aromatic feature: while the inner eight-membered ring is anti-aromatic, the outer fused rings are aromatic.The predicted UV spectrum and reduction potential of 1a agree well with available experimental values. Following replacement of H-atoms by F-atoms, the energy levels of frontier orbitals of 1b–4b are consistently decreased as compared to those of 1a–4a.
Based on the properties of electrochemistry and charge transport calculated, the molecules 1a–4a and 4b can be regarded as good candidates for p-type semiconductors. More importantly, the molecules 1b–3b are revealed to exhibit characteristics suitable for ambipolar organic semiconductors. Since the n-type and ambipolar organic semiconducting materials remain rare, we would hope that the present results will attract much attention from the experimental scientists.
At the time of finalizing this report, we interestingly found that tetrathio and tetraselono[8]circulenes were experimentally synthesized by Wong et al.65
Acknowledgements
We are indebted to the KU Leuven Research Council (GOA) and Vlaams Supercomputer Centrum (VSC). VTTH and TBT would like to thank the FWO-Vlaanderen for Ph. D. and postdoctoral fellowships.Notes and references
- W. E. Barth and R. E. Lawton, J. Am. Chem. Soc., 1966, 88, 380–381 CrossRef CAS.
- R. Scholl and K. Meyer, Ber. Dtsch. Chem. Ges. A, 1932, 65, 902–915 CrossRef.
- K. Yamamoto, T. Harada, M. Nakazaki, Y. Naka, Y. Kai, S. Harada and N. Kasai, J. Am. Chem. Soc., 1983, 105, 7171–7172 CrossRef CAS.
- R. Salcedoa, L. E. Sansoresa, A. Picazoa and L. Sansón, J. Mol. Struct.: THEOCHEM, 2004, 678, 211–215 CrossRef.
- C. N. Feng, M. Y. Kuo and Y. T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791–7794 CrossRef CAS.
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367 CrossRef CAS PubMed.
- A. Dadvand, F. Cicoira, Y. K. Chernichenko, E. S. Balenkova, R. M. Osuna, F. Rosei, V. G. Nenajdenko and D. F. Perepichka, Chem. Commun., 2008, 5354–5356 RSC.
- A. Datta and S. K. Pati, J. Phys. Chem. C, 2007, 19, 4487–4490 Search PubMed.
- S. Mohakud and S. K. Pati, J. Mater. Chem., 2009, 19, 4356–4361 RSC.
- G. Gahungu, J. Zhang and T. Barancira, J. Phys. Chem. A, 2009, 113, 255–262 CrossRef CAS PubMed.
- X. D. Tang, Y. Liao, H. Z. Gao, Y. Geng and Z. M. Su, J. Mater. Chem., 2012, 22, 6907–6918 RSC.
- V. T. T. Huong, T. B. Tai and M. T. Nguyen, Phys. Chem. Chem. Phys., 2012, 14, 14832–14841 RSC.
- T. K. Mandal, D. Jose, A. Nijmudheen and A. Datta, J. Phys. Chem. C, 2014, 118, 12115–12120 CAS.
- T. B. Tai, V. T. T. Huong and M. T. Nguyen, Chem. Commun., 2013, 49, 11548–11550 RSC.
- H. Erdtman and H. E. Högberg, Chem. Commun., 1968, 773–774 RSC.
- (a) B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Comput. Theor. Chem., 2011, 972, 68–74 CrossRef CAS; (b) V. A. Minaeva, B. F. Minaev, G. V. Baryshnikov, H. Agren and M. Pittelknow, Vib. Spectrosc., 2012, 61, 156–166 CrossRef CAS; (c) N. N. Karaush, B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Opt. Spectrosc., 2014, 116, 37–51 CrossRef; (d) V. A. Minaeva, B. F. Minaev, G. V. Baryshnikov, O. A. Romeyko and M. Pittelknow, Vib. Spectrosc., 2013, 65, 147–158 CrossRef CAS; (e) G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva, A. T. Baryshnikova and M. Pittelknow, J. Mol. Struct., 2012, 1026, 127–132 CrossRef CAS.
- (a) G. V. Baryshnikov, R. R. Valiev, N. N. Karaush and B. F. Minaev, Phys. Chem. Chem. Phys., 2014, 16, 15367–15374 RSC; (b) G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva and V. G. Nenajdenko, J. Mol. Model., 2013, 19, 4511–4519 CrossRef CAS PubMed.
- C. B. Nielsen, T. Brock-Nannestad, T. K. Reenberg, P. Hammershøj, J. B. Christensen, J. W. Stouwdam and M. Pittelkow, Chem.–Eur. J., 2010, 16, 13030–13034 CrossRef CAS PubMed.
- (a) C. B. Nielsen, T. Brock-Nannestad, P. Hammershoj, T. K. Reenberg, M. S. Magnussen, D. Trpcevski, T. Hessel, R. Salcado, G. V. Baryshnikov, B. F. Minaev and M. Pittelkow, Chem.–Eur. J., 2013, 19, 3898–3904 CrossRef CAS PubMed; (b) T. Hensel, D. Trpcevski, C. Lind, R. Grosjean, P. Hammershoj, C. B. Nielsen, M. S. Magnussen, B. Minaev, G. V. Baryshnikov and M. Pittelkow, Chem.–Eur. J., 2013, 19, 17097–17102 CrossRef CAS PubMed; (c) V. A. Minaeva, B. F. Minaev, G. V. Baryshnikov and M. Pittelkow, Opt. Spectrosc., 2013, 114, 509–512 CrossRef CAS.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, Phys. Chem. Chem. Phys., 2014, 16, 6555–6559 RSC.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. Minaeva, RSC Adv., 2014, 4, 25843–25851 RSC.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef.
- M. M. Francl, W. J. Petro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- G. W. Trucks, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. F. Nelsen and F. Blomgren, J. Org. Chem., 2001, 66, 6551–6559 CrossRef CAS PubMed.
- S. F. Nelsen, D. A. Trieberi, R. F. Ismagilov and Y. Teki, J. Am. Chem. Soc., 2001, 123, 5684–5694 CrossRef CAS PubMed.
- M. Malagoli and J. L. Brédas, Chem. Phys. Lett., 2000, 327, 13–17 CrossRef CAS.
- K. Sakanoue, M. Motoda, M. Sugimoto and S. Sakaki, J. Phys. Chem. A, 1999, 103, 5551–5556 CrossRef CAS.
- X. Y. Li, J. Tong and F. C. He, Chem. Phys., 2000, 260, 283–294 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- A. Troisi and G. Orlandi, Chem. Phys. Lett., 2001, 344, 509–518 CrossRef CAS.
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 CrossRef CAS PubMed.
- S. Yin, Y. Yi, Q. Li, G. Yu, Y. Liu and Z. Shuai, J. Phys. Chem. A, 2006, 110, 7138–7143 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS.
- J. Huang and M. Kertesz, Chem. Phys. Lett., 2004, 390, 110–115 CrossRef CAS.
- H. Li, R. Zheng and Q. Shi, J. Phys. Chem. C, 2012, 116, 11886–11894 CAS.
- X. Yang, L. Wang, C. Wang, W. Long and Z. Shuai, Chem. Mater., 2008, 20, 3205–3211 CrossRef CAS.
- G. Nan and Z. Li, Org. Electron., 2012, 13, 1229–1236 CrossRef CAS.
- X. Yang, Q. Li and Z. Shuai, Nanotechnology, 2007, 18, 424029 CrossRef PubMed.
- V. T. T. Huong, T. B. Tai and M. T. Nguyen, Phys. Chem. Chem. Phys., 2012, 14, 14832–14841 RSC.
- X. D. Tang, Y. Liao, H. Z. Gao, Y. Geng and Z. M. Su, J. Mater. Chem., 2012, 22, 6907–6918 RSC.
- X. K. Chen, L. Y. Zou, J. F. Gou and A. M. Ren, J. Mater. Chem., 2012, 22, 6471–6484 RSC.
- (a) J. Huang and M. Kertesz, Chem. Phys. Lett., 2004, 390, 110–115 CrossRef CAS; (b) C. Sutton, J. S. Sears, V. Coropceanu and J.-L. Bredas, J. Phys. Chem. Lett., 2013, 4, 919 CrossRef CAS PubMed.
- A. P. Davis and A. J. Fry, J. Phys. Chem. A, 2010, 114, 12299–12304 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Namazian, C. Y. Lin and M. L. Coote, J. Chem. Theory Comput., 2010, 6, 2721–2725 CrossRef CAS.
- J. E. Bartmess, J. Phys. Chem., 1994, 98, 6420–6424 CrossRef CAS.
- G. Nan, X. Yang, L. Wang, Z. Shuai and Y. Zhao, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115203 CrossRef.
- S. Chai, S. H. Wen, J. D. Huang and K. L. Han, J. Comput. Chem., 2011, 32, 3218–3225 CrossRef CAS PubMed.
- (a) G. Gahungu, B. Zhang and J. P. Zhang, J. Phys. Chem. C, 2007, 111, 4838–4846 CrossRef CAS; (b) G. Gahungu and J. Zhang, Phys. Chem. Chem. Phys., 2008, 10, 1743–1747 RSC.
- C. Wartelle, R. Viruela, P. M. Viruela, F. X. Sauvage, M. Salle, E. Orti, E. Levillain and F. Le Derf, Phys. Chem. Chem. Phys., 2003, 5, 4672–4679 RSC.
- E. G. Kim, V. Coropceanu, N. E. Gruhn, R. S. Sanchez-Carrera, R. Snoeberger, A. J. Matzger and J. L. Bredas, J. Am. Chem. Soc., 2007, 129, 13072–13081 CrossRef CAS PubMed.
- S. T. Bromley, M. Mas-Torrent, P. Hadley and C. Rovira, J. Am. Chem. Soc., 2004, 126, 6544–6545 CrossRef CAS PubMed.
- P. v. R. Schleyer, H. Jiao, N. J. R. V. E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- T. B. Tai, N. M. Tam and M. T. Nguyen, Theor. Chem. Acc., 2012, 131, 1241 CrossRef and references therein.
- J. Andzelm, B. C. Rinderspacher, A. Rawlett, J. Dougherty, R. Baer and N. Govind, J. Chem. Theory Comput., 2009, 5, 2835–2846 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. J. Perpete and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS.
- C. R. Newman, C. D. Frisbie, D. A. S. Filho, J. L. Bredas, P. C. Ewbank and K. P. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS and references therein.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- X. Xiong, C. L. Deng, B. F. Minaev, G. V. Baryshnikov, Z. S. Peng and H. N. C. Wong, Chem.–Asian J., 2015 DOI:10.1002/asia.201403028.
Footnote |
| † Electronic supplementary information (ESI) available: The xyz-coordinates of the optimized structures 1a–4a and 1b–4b at the neutral state. See DOI: 10.1039/c4ra16485g |
| This journal is © The Royal Society of Chemistry 2015 |