DOI:
10.1039/C4RA16460A
(Paper)
RSC Adv., 2015,
5, 13315-13323
Polydopamine-derived porous nanofibers as host of ZnFe2O4 nanoneedles: towards high-performance anodes for lithium-ion batteries†
Received
16th December 2014
, Accepted 19th January 2015
First published on 19th January 2015
Abstract
In this work, highly mesoporous carbon nanofibers in free-standing mat form are successfully fabricated by single-spinneret electrospinning of polystyrene (PS) followed by coating the porous PS nanofibers via in situ polymerization of dopamine and subsequent annealing. The pores inside the nanofibers are mainly in the range of 10–50 nm and interconnected to each other, forming nanochannels. ZnFe2O4 crystals can then be grown from the nanofibers via a solution route. Strikingly, ZnFe2O4 nanoneedles are formed, which have diameter and length of about 8 nm and 70 nm, respectively, and are located evenly not only on the surface of the nanofibers but also inside the nanochannels. The ZnFe2O4/carbon composite nanofibers exhibit excellent cyclability and rate performance as anodes of lithium ion batteries (LIBs), in which the ZnFe2O4 nanoneedles are the major active component with normalized capacity of 1000–1700 mA h g−1 at 0.1 A g−1 and 560 mA h g−1 at 5 A g−1, respectively. The excellent properties can be ascribed to the very small diameter of the nanoneedles that ensures complete conversion reactions and alloying/de-alloying between Zn and lithium, the good contact of the nanoneedles with polydopamine-derived N-doped graphitic carbon that offer efficient electrical conduction, and the nanochannels that allow facile transport of the electrolyte and lithium ions.
Introduction
Carbonaceous materials have been used as lithium-ion battery (LIB) anodes for a few decades.1–4 Concerning energy storage for high-power tools and electrical vehicles, the relatively low theoretical specific capacity of the current commercial LIB anode material, graphite, is one of the key technical barriers.5,6 Metal oxides, in particular binary/polynary ones such as Li4Ti5O12,7 ZnM2O4 (M = Co, Mn, etc.),8,9 and MFe2O4 (M = Cd, Ni, Co, Zn, etc.),10–13 have emerged as promising alternatives due to their much higher theoretical capacities. Among these, zinc ferrite (ZnFe2O4) has received extensive attention due to its low cost and high safety,14 as well as the extra lithium uptake by Li–Zn alloy.15,16 However, the poor electrical conductivity as well as large volume expansion-induced capacity decay hinders its practical applications as LIB anode materials. Many strategies have been developed to address these issues. Typical ones include fabricating nanometer-sized ZnFe2O4,14,17,18 introducing pores into the electrodes,19,20 and combining ZnFe2O4 with semiconductors,21,22 metallic dopants,23,24 or conductive phases, especially carbon.25–28 Improved electrochemical performance was achieved compared with the raw, bulky ZnFe2O4. As typical examples, carbon-coated ZnFe2O4 nanoparticles show cycled capacity of 1000–1400 mA h g−1 at 0.04 A g−1 and about 500 mA h g−1 at 3.89 A g−1;27 while yolk–shell structured ZnFe2O4 powders deliver cycled capacity of 700–900 mA h g−1 at 0.5 A g−1.18
The introduction of the high-capacity materials into carbon matrices, including carbon powder, nanospheres, nanofibers, nanotubes as well as graphene/graphite, indeed enhances cyclability and rate capacity of the anodes through strengthening charge conduction and stabilizing anode structures.29–33 Besides, porous carbon34,35 can provide additional benefits because the specific surface area of the porous carbon is larger than that of the solid ones, leading to larger contact area and hence potentially more efficient interactions between the electrodes and electrolytes, and the pores also provide open channels for transport of lithium ions through electrolyte. Porous carbon also allows the growth of micron- or nanometer-sized high-capacity materials inside the pores,36 creating favorable interactions between the two phases.
To maximize the functions of porous carbon, its structures, such as the contents of graphitic carbon and heteroatom dopants, and morphology, such as the pore size and volume, need to be carefully controlled. In our previous publications, we have reported that polydopamine (PDA) is an excellent carbon source for LIB applications.31,36,37 Carbonized PDA (C-PDA) has multi-layered graphene-like structure doped with high contents of pyridinic and pyrrolic N, and its in-plane and trough-plane electrical conductivities are comparable to that of multi-layered graphene.37 Furthermore, PDA, a synthetic mimic of an essential component of mussel adhesive proteins, can well adhere to a wide variety of organic and inorganic surfaces.38 Thus, by self-polymerization of dopamine in an alkaline aqueous solution, PDA can be facilely coated onto polymer nanofibers with controllable thickness. This opens up the possibility to use porous polymer nanofibers as the sacrificing template to create porous C-PDA nanofibers with tailored morphology, which can then act as a highly conductive host for nano-sized high-capacity materials. In this work, free-standing mesoporous carbon nanofibers with interpenetrating nanochannels were prepared by facile in situ coating of PDA on electrospun porous polystyrene nanofibers and subsequent carbonization. ZnFe2O4 nanoneedles were then grown from the mesoporous carbon nanofibers via a solution route. Herein we demonstrate that by synergistic combination of the unique C-PDA structures with the mesoporous morphology of the nanofibers and nanoneedle-like morphology of ZnFe2O4, the obtained nanocomposites exhibit excellent rate performance and cyclability as LIB anodes.
Experimental
Materials
Polystyrene (PS, Mw = 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), dopamine hydrochloride (DOPA), tris(hydroxymethyl)aminomethane (Tris) were purchased from Sigma-Aldrich Chemistry (USA). Zinc sulfate heptahydrate (ZnSO4·7H2O, ≥99.0%), iron sulfate heptahydrate (FeSO4·7H2O, ≥99.0%) and ammonium hydroxide (NH3·H2O, 28.0–30.0% NH3 basis) were purchased from Aldrich (USA). Dimethylformamide (DMF) was purchased from Tedia Company Inc (USA). Ethanol was supplied by Merck KGaA (Germany). All materials were used without further purification.
000), dopamine hydrochloride (DOPA), tris(hydroxymethyl)aminomethane (Tris) were purchased from Sigma-Aldrich Chemistry (USA). Zinc sulfate heptahydrate (ZnSO4·7H2O, ≥99.0%), iron sulfate heptahydrate (FeSO4·7H2O, ≥99.0%) and ammonium hydroxide (NH3·H2O, 28.0–30.0% NH3 basis) were purchased from Aldrich (USA). Dimethylformamide (DMF) was purchased from Tedia Company Inc (USA). Ethanol was supplied by Merck KGaA (Germany). All materials were used without further purification.
Preparation of carbonized polydopamine (C-PDA) nanofibers
PS was firstly dissolved in DMF under magnetic stirring at 60 °C to form PS–DMF solution with PS concentration of 15 wt%. 2.5 mL PS–DMF solution was electrospun into PS nanofibers at the feeding rate of 0.5 mL h−1 and under the working voltage of 12 kV. Ethanol was used as liquid collector as reported.36 The PS nanofibers were then transferred into 600 g DOPA aqueous solution (0.3 mg mL−1). The coating of PS nanofibers was carried out by triggering the in situ polymerization of DOPA via adding 0.73 g Tris into the above aqueous solution under continuous shaking. The coating time was 2 h. The above coating procedure was repeated for another 1–5 times using fresh DOPA solution to achieve a series of coating thickness. After being washed by de-ionized water (DI H2O) and freeze-dried, the PDA-coated PS nanofibers were annealed in argon environment at 700 °C for 3 h. The annealed samples are denoted as PNFs-1, PNFs-2, PNFs-3, PNFs-4, PNFs-5 and PNFs-6, respectively, in which the number specifies how many times of PDA coating that have been conducted.
Growth of ZnFe2O4 nanoneedles in porous C-PDA nanofibers
The precursor solution was firstly prepared by dissolving ZnSO4·7H2O and FeSO4·7H2O into 100 mL DI H2O with concentration of 0.1 mmol and 0.2 mmol, respectively. Two typical samples, PNFs-2 and PNFs-6, were chosen for hosting ZnFe2O4. Briefly, the sample was immersed in the precursor solution for 6 h and then horizontally suspended above 50 mL concentrated ammonium hydroxide for 24 h in a sealed glass bottle. The treated sample was washed by DI H2O for 3 times, dried in vacuum at 60 °C and finally annealed. The annealing condition was as following: heating to 300 °C in atmosphere at the heating rate of 5 °C min−1, keeping at 300 °C for 3 h, and then further heating to 600 °C in argon flow at the heating rate of 5 °C min−1 and keeping at 600 °C for another 3 h. The annealed samples were denoted as ZnFe–PNFs-2 and ZnFe–PNFs-6, respectively.
Structural, morphological and electrochemical characterization
Morphologies of the samples were investigated using a field-emission scanning electron microscope (FESEM, JEOL-7600F) and a transmission electron microscope (TEM, JEOL-2100F). For cross-sectional observation, the samples were embedded into epoxy, cut into ultrathin slides using a TEM ultramicrotome and deposited onto copper grid. The Brunauer–Emmett–Teller (BET) specific surface area and Barrett–Joyner–Halenda (BJH) pore size of the samples were measured using a Micromeritics Tristar II-3020 nitrogen adsorption apparatus. The structure and composition of the nanocomposite samples were studied using an X-ray diffractometer (XRD, Bruker D8 Discover GADDS) and a thermogravimetric analyzer (TGA, Q500), respectively. Cyclic voltammetry (CV) tests were performed on an electrochemical workstation (Autolab, PGSTAT302). For battery tests, the free-standing mat of ZnFe–PNFs-6 (or PNFs-6) was used as the anode directly through punching into round shape of 10 mm in diameter, while lithium foil (Sigma Chemicals, USA) and 1 M LiPF6 in the mixture of ethylene carbonate and dimethyl carbonate with 1/1 volumetric ratio (Charslton Technologies, Singapore) were used as the counter electrode and electrolyte, respectively. The three components were assembled into button cells (CR2032, ACME, Singapore) using a separator (Celgard2325, USA) as reported previously.30 The cycled and rate capacity were measured using a battery test system (MACCOR, 4200).
Results and discussion
Preparation and morphology of the porous C-PDA nanofibers
The porous C-PDA nanofibers were prepared by coating PDA onto electrospun porous PS nanofibers followed by annealing in argon environment. Uniform PS nanofibers with average diameter of around 830 nm were achieved via electrospinning of PS–DMF solution (Fig. 1a). The pore structure of the electrospun nanofibers is crucially affected by the environmental humidity and solvent property.39 Using DMF as the solvent in this case, the PS was electrospun into highly porous nanofibers (inset of Fig. 1a) under the environmental humidity of about 50%. The pores are created across each nanofiber, forming interconnected nanochannels (Fig. 1b).
 |
| | Fig. 1 Morphology ((a) side view, (b) cross-sectional view) of porous polystyrene nanofibers by electrospinning of PS–DMF solution at environmental humidity of about 50%. The inset in (a) shows the surface feature of a single nanofiber. | |
PDA was coated onto the porous PS nanofibers via in situ polymerization of DOPA in slight alkaline environment. Owing to the good affinity of PDA to polystyrene,38 the coating is uniformly formed on the PS nanofibers. A low concentration of DOPA, only 0.3 mg mL−1, was used to, firstly, avoid the formation of free PDA particles, and secondly, to slow down the polymerization speed in order to deposit PDA on the surface of the pores located inside the PS nanofibers; this is possible because the dissolved DOPA can easily diffuse into the nanochannels through the open pores on the surface of the PS porous nanofibers. After annealing at 700 °C in argon flow, PS is fully removed via degradation,36 while PDA is converted to carbon, as confirmed by TGA results (Fig. S1†). The derived carbon has layered structure and is doped nitrogen and possesses comparable electrical conductivity to that of multi-layered graphene.37 C-PDA also contains ∼4.5% nitrogen (N), which is mainly composed of pyridinic (35.2%) and pyrrolic (52.4%) N. The heteroatoms lead to structural disorder and distortion that may favor reversible lithium storage, which will be further elaborated later. By controlling PDA coating thickness, two different types of porous morphologies, as shown in Scheme 1, are achieved after the annealing.
 |
| | Scheme 1 A schematic showing cross-sectional views of C-PDA porous nanofibers with different morphologies. | |
The morphology of the porous carbon nanofibers was investigated using SEM and TEM. The nanofibrous morphology is well retained after removing the PS template regardless of the PDA coating thickness (Fig. 2a1–f1). The nanofibers are highly porous (Fig. 2a3–f3), resulting from the template effect of the porous nanofibers. From the high-magnification FESEM images (Fig. 2a2–f2), it can be seen that the surface of the C-PDA nanofibers becomes smoother with thicker coatings. The PDA coating thicknesses of PNFs-1, PNFs-2 and PNFs-3 are smaller than the size of the pores in the PS nanofibers. For instance, the final C-PDA wall thickness is only about 7 nm for PNFs-2 as measured from the TEM images (Fig. 2b3), while the original pore openings on the porous PS nanofiber surface have diameter of tens of nanometers (Fig. 1a inset). Therefore the pore openings on the PS nanofiber surface are probably not completely blocked by the PDA coating, giving rough C-PDA surfaces after the removal of PS. In this case, the open pores would allow efficient medium exchange for further use (Fig. 2a3–c3). The C-PDA wall thickness is increased to around 27 nm for PNFs-6, implying that the much thicker PDA coating blocked the pore openings on the PS nanofiber surface and therefore giving smoother nanofiber surface after removing PS (Fig. 2d3–f3). The porosity as well as surface features of the nanofibers are further confirmed by TEM cross-sectional views (Fig. 2a4–f4), where rough surfaces with open channels can be clearly seen (indicated by the red dash arrows in Fig. 2a4–c4) for PNFs-1, PNFs-2 and PNFs-3, while relatively dense C-PDA shells are distinct for PNFs-5 and PNFs-6. PNFs-2 with obvious open pores on surface and PNFs-6 with a denser outer shell were taken as typical examples for BET measurements. Fig. 3 shows that the BET specific surface area of the two samples are about 306 m2 g−1 and 507 m2 g−1, respectively, while the BJH desorption pore size is mainly in the range of 10–50 nm for both. The increase of the surface area from PNFs-2 to PNFs-6 is probably due to the extra surface created by the formed continuous shell in PNFs-6 as well as the presence of more large pores of PNFs-2, which are beyond the measuring limit of the equipment.
 |
| | Fig. 2 Porous C-PDA nanofibers with PDA coating time(s) of (a1–a4) 1 (PNFs-1), (b1–b4) 2 (PNFs-2), (c1–c4) 3 (PNFs-3), (d1–d4) 4 (PNFs-4), (e1–e4) 5 (PNFs-5) and (f1–f4) 6 (PNFs-6). | |
 |
| | Fig. 3 (a) BET adsorption/desorption isotherm and (b) BJH desorption pore size distribution of C-PDA nanofibers (PNFs-2 and PNFs-6). | |
The highly porous C-PDA nanofibers obtained are ideal host for trapping/growth of other active materials due to their high porosity, proper pore size, stable carbonaceous microstructure and the free-standing nature of the nanofibrous mats, as well as high electrical conductivity of C-PDA.37 In addition, by tailoring the surface feature of the nanofibers, the nanofibers may be used for different applications. Based on the microscopic studies, the porous C-PDA nanofibers are classified into two categories: the ones with obvious open pores on surface and that with relatively dense outer shells. The former offers numerous channels, including the open pores on the surface and at the two ends of the nanofibers, to facilitate fast mass/charge transport. Differently, the latter allows liquid medium infusion/diffusion/transport mainly from the two ends of the nanofibers since the surface does not have large open pores. In this case, the relatively dense shell may act as a barrier layer to slow down the leaching of the trapped fluids, facilitating the growth of crystals in the nanofibers.
Growth of ZnFe2O4 nanoneedles in porous C-PDA nanofibers
Two typical samples, PNFs-2 and PNFs-6, were used to host ZnFe2O4, which is a promising anode material for LIBs as mentioned in the introduction part. The introduction of ZnFe2O4 into the C-PDA porous nanofibers was achieved via a solution route (Scheme 2). After PNFs-2 and PNFs-6 being immersed into the aqueous solution of ZnSO4·7H2O–FeSO4·7H2O for a few hours and taken out, the precursors are trapped inside the nanofibers. The two hosts have different trapping capability due to their difference in morphology. For PNFs-2, the open pores on the surface do not effectively hold the precursor solution in the nanofibers, leading to very low ZnFe2O4 content in the final product (6.4 wt%, Fig. S2†). By contrast, the relatively dense shell of PNFs-6 effectively protects the infused precursor solution from outflowing. Additionally, some solution is also adsorbed onto the shell, forming a thin precursor layer on the outer surface. When the samples with the precursor are horizontally suspended above the concentrated NH3·H2O solution in an enclosed environment, the NH3 steam diffuses into the C-PDA porous nanofibers and converts the trapped ZnSO4·7H2O and FeSO4·7H2O into zinc complexes with hydroxide or ammonia40 and Fe(OH)3, respectively, in the presence of oxygen. After being oxidized at 300 °C in air and further annealed in argon atmosphere at 600 °C, the intermediate is converted into ZnFe2O4. The formation of ZnFe2O4 is confirmed by the XRD pattern shown in Fig. 4a. The intensive peaks at around 25°, 18.5°, 30.3°, 35.5°, 38.0°, 43.5°, 51.4°, 57.4° and 63.0° can be assigned to diffraction plane [002] of graphitic C-PDA, and [111], [220], [311], [222], [400], [422], [333] and [440] plane of ZnFe2O4 in cubic structure, respectively, based on JCPDS no. 22-1012. No impurity peaks are observed. The existence of Zn and Fe in ZnFe2O4 is further confirmed by the STEM mapping results (Fig. 4b). The mapping results also show that the distributions of both Zn and Fe form nanoneedle shape, which will be elaborated later. The calculated inter-plane d spacings for [111], [220], [311], [222] and [400] of ZnFe2O4 are 0.48 nm, 0.29 nm, 0.25 nm, 0.24 nm and 0.21 nm, respectively. The content of ZnFe2O4 in ZnFe–PNFs-6 is about 33.2 wt% based on TGA (Fig. 4c).
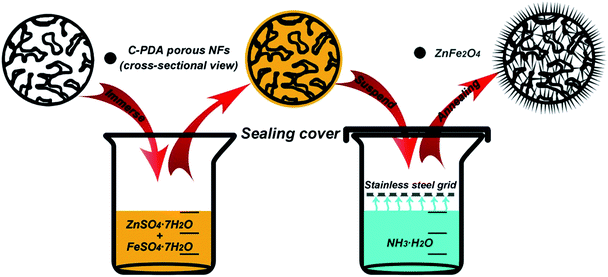 |
| | Scheme 2 A schematic showing preparation procedure for the growth of ZnFe2O4 nanoneedles in C-PDA porous nanofibers. | |
 |
| | Fig. 4 (a) XRD pattern, (b1–b3) STEM elemental mapping results of Zn and Fe and (c) TGA curve of ZnFe–PNFs-6. | |
Instead of forming nanoparticles,13,17,25–27,41–44 ZnFe2O4 nanoneedles are formed uniformly on the surface of the C-PDA porous nanofibers (Fig. 5a and b). The morphological studies of the samples before annealing (Fig. S3†) reveal that the nanoneedles have already been formed after ammonia treatment. The ZnFe2O4 nanoneedles with length and diameter of about 70 nm and 8 nm, respectively, mainly align perpendicularly to the nanofibers surface (Fig. 5c). The alignment indicates that the nanoneedles are formed upon nucleation on the C-PDA surfaces. The size of the nucleation sites may be restricted by the mesoporous nature of the nanofibers, which is probably the reason for the very small lateral size of the ZnFe2O4 crystals. The growth direction, which is indicated by the green arrows in Fig. 5d1 and d2, is along [111] plane with inter-planar spacing of 0.48 nm. The high-resolution TEM image (Fig. 5d2) also verify the single-crystalline nature of each individual nanoneedle, where the inter-planar spacing for plane [200], [311] and [222] is 0.41 nm, 0.25 nm and 0.24 nm, respectively.14 This is consistent with the calculated d spacings from XRD pattern shown above. The TEM cross-sectional images, as shown in Fig. 5e and f, indicate that the nanoneedles are formed not only on the surface of nanofibers but also inside the pores. Since the alignment is perpendicular to the surface as verified above, the length of these nanoneedles is restricted by the size of the pores that are in the ranges of 10–50 nm. This is also confirmed by the TEM image taken from the center area of the cross-section (Fig. 5f), where randomly aligned nanoneedles can be clearly observed.
 |
| | Fig. 5 (a and b) The morphology of PNFs-6 after growth of ZnFe2O4 nanoneedles. (c) The morphology and (d) crystal structure of the ZnFe2O4 nanoneedles. The green arrows in image (d1) and (d2) indicate the growth (length) direction of the nanoneedles. (e and f) Cross-sectional views of a single porous nanofiber with the ZnFe2O4 nanoneedles. | |
Electrochemical performance of the porous composite nanofibers
The electrochemical properties of the free-standing ZnFe2O4 nanoneedles-in-C-PDA nanofibers, ZnFe–PNFs-6, as a LIB anode were investigated because it has higher ZnFe2O4 content than other samples. The CV curves for the 1st–5th cycle (Fig. 6a) show typical lithiation/de-lithiation behavior of carbon/ZnFe2O4-based composites. The weak cathodic peak at about 1.5 V in the 1st cycle can be assigned to the irreversible reaction between lithium ions and electrolyte, forming solid electrolyte interface (SEI), and the peak disappears in the following cycles.45 A weak shoulder at about 0.9 V and a intensive peak at 0.64 V on the 1st cathodic curve are ascribed to the lithiation of ZnFe2O4 (eqn (1)), which gives metallic Zn and Fe irreversibly, as well as the formation of Li–Zn alloy (eqn (2)).16| | |
ZnFe2O4 + 8Li+ + 8e− → Zn + 2Fe + 4Li2O
| (1) |
 |
| | Fig. 6 (a) Cyclic voltammetry (CV) curves (1st–5th cycle, the inset shows the enlarged anodic profile), (b) discharge–charge curves (1st–5th cycle), (c) cycled capacity at 0.05 A g−1 followed by 0.1 A g−1, (d) rate capacity and (e) cycled capacity at 1 A g−1 of ZnFe–PNFs-6. The blue and green lines in (c) and (d) show the cycled and rate capacity of pure PNFs-6 and the normalized cycled and rate capacity of pure ZnFe2O4 nanoneedles, respectively, indicating the contribution of pure PNFs-6 and ZnFe2O4 nanoneedles to the overall anode performance. | |
Noting that the carbon content in this nanostructure is as high as 66.8 wt%, and hence the porous C-PDA nanofibers may also contribute to the overall electrochemical performance of the anode. This is confirmed by the partially overlapped peak at 0.55 V and the sharp slop at less than 0.3 V, which are due to the lithiation of C-PDA,36 and will be further clarified later. Several partially overlapped obvious humps (centred at about 0.4 V, 1.2 V and 1.6 V, respectively) within the range of 0.2–2.0 V on the 1st anodic curve, which are also observed in the following anodic curves, correspond to the de-lithiation of C-PDA (around 1.2 V),36 the de-alloying of LiZn (eqn (3))46 and the oxidation of metallic Zn and Fe with formed Li2O (eqn (4) and (5)).13,17
| | |
LiZn → Zn + Li+ + e− (around 0.4 V)
| (3) |
| | |
Zn + Li2O → ZnO + 2Li+ + 2e− (around 1.6 V)
| (4) |
| | |
2Fe + 3Li2O → Fe2O3 + 6Li+ + 6e− (around 1.6 V)
| (5) |
In the subsequent cathodic cycles, the intensive peak at about 0.8 V is assigned to the reversible reduction of ZnO and Fe2O3 (in reverse direction of eqn (4) and (5)),13 while the slop that starts at around 0.6 V is relative to the Li–Zn alloying46 followed by C-PDA lithiation.36 Therefore the reversible capacity involves both the Zn–Li2O–ZnO and Fe–Li2O–Fe2O3 conversions as well as the Li–Zn alloying/de-alloying and C-PDA lithiation. The constant peak intensity from the 2nd cycle onwards indicates that the capacity of the anode can be well maintained.
As shown in Fig. 6b, the anode delivers initial discharge and charge capacity of 1581 and 958 mA h g−1, respectively, with initial coulombic efficiency of about 61% at specific current of 0.05 A g−1 within 3.0–0.005 V. It is noted that the galvanostatic discharge–charge profiles are fairly similar to that of neat porous C-PDA nanofibers (PNFs-6, Fig. S4†) rather than ZnFe2O4.16 This is due to the higher content of carbon (66.8 wt%) than that of the ZnFe2O4 nanoneedles (33.2 wt%). The low initial coulombic efficiency is mainly due to the lithiation of ZnFe2O4 during the first discharge, as shown in eqn (1), that irreversibly consume extra lithium as well as the formation of SEI. The capacity quickly stabilizes in the subsequent cycles. At a higher specific current of 0.1 A g−1, the capacity drops to around 700 mA h g−1 initially, then gradually increases and stabilizes at about 900 mA h g−1 (Fig. 6c, red line). Further increase of the specific current reduces the reversible capacity of the anode (Fig. 6d, red line). Nevertheless, at a high specific current of 5 A g−1, a capacity value of about 300 mA h g−1 can still be achieved. The capacity fully rebounds back to 900 mA h g−1 when the specific current is reduced back to 0.1 A g−1. The same battery was further cycled at 1 A g−1 to test its lifetime. As shown in Fig. 6e, the reversible capacity stabilizes at around 700 mA h g−1 for over 200 cycles, indicating the excellent cyclability and long lifetime of the composite nanofibers. The excellent electrochemical performance is ascribed to the integrated effect of both C-PDA porous nanofibers and ZnFe2O4 nanoneedles. In general the C-PDA porous nanofibers can ensure good charge conduction, resistance-free pathways for transport of ions and electrolyte, as well as provide some capacity, while the ZnFe2O4 nanoneedles with such a small diameter may be able to ensure complete conversions and alloying/de-alloying with lithium ions. In order to further clarify the role played by each component, the cycled and rate capacity of C-PDA porous nanofibers, PNFs-6, was measured under the same conditions as that of ZnFe–PNFs-6 and presented as blue lines in Fig. 6c and d. It is shown that PNFs-6 delivers reversible capacity of around 600 mA h g−1 and 480 mA h g−1 at 0.05 A g−1 and 0.1 A g−1, respectively. The capacity decreases to about 75 mA h g−1 and 15 mA h g−1 at high specific current of 5 A g−1 and 10 A g−1, respectively. Due to the considerable amount of doped pyridinic and pyrrolic N and induced defects, the major lithiation/de-lithiation process of C-PDA is metallic lithium plating that induced by defects instead of standard stage I formation/extraction of LiC6 intercalates.36,47,48 This is confirmed by its galvanostatic discharge–charge profiles (Fig. S4†). Assuming that the overall anode capacity is the sum of both C-PDA and ZnFe2O4, the contribution of ZnFe2O4 nanoneedles to the overall performance can be evaluated as normalized capacity of ZnFe2O4, which is calculated from the following formula:
| CapZnFe2O4 = (Capanode − Capcarbon × Ccarbon)/CZnFe2O4 |
where Cap
ZnFe2O4, Cap
anode and Cap
carbon represent normalized capacity of ZnFe
2O
4 nanoneedles, measured capacity of anode and porous carbon nanofibers (PNFs-6), respectively, while
Ccarbon and
CZnFe2O4 are the contents of PNFs-6 (66.8 wt%) and ZnFe
2O
4 nanoneedles (33.2 wt%), respectively. As presented by the green lines in
Fig. 6c and d, the ZnFe
2O
4 nanoneedles possess normalized cycled capacity of 1000–1700 mA h g
−1 at a specific current of 0.1 A g
−1. This suggests the full lithiation/de-lithiation of ZnFe
2O
4 nanoneedles during the cycling, taking note that the theoretical capacity of ZnFe
2O
4 is 1072 mA h g
−1.
22 The normalized capacity at high specific current of 5 A g
−1 is still about 560 mA h g
−1, more than 50% of the theoretical value. The capacity performance of the ZnFe
2O
4 nanoneedles is comparable or even superior to that of reported ZnFe
2O
4-based anodes as presented in introduction section, indicating their significant contribution to the overall electrochemical performance of the composite nanofibers.
Conclusions
Highly mesoporous carbon nanofibers in free-standing mat form were successfully prepared by single-spinneret electrospinning of porous PS nanofibers followed by in situ polymerization of dopamine and annealing. The pores are mainly of 10–50 nm in size and interconnected to each other, forming nanochannels. The surface of the nanofibers has either open pores or be covered by a relatively dense shell, depending on the overall PDA coating thickness. ZnFe2O4 nanoneedles can grow from the mesoporous carbon nanofibers via the solution route. The ZnFe2O4 nanoneedles are located on the surface as well as inside the nanofibers. The nanoneedle-like morphology of ZnFe2O4, the efficient contact between the nanoneedles and highly conductive C-PDA matrix, and the facile diffusion/transport of electrolyte and charges in the nanochannels lead to excellent cyclability and rate capacity for the composite nanofiber anode, where the ZnFe2O4 nanoneedles are the major contributor to the overall performance.
Notes and references
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Small, 2013, 9, 1173 CrossRef CAS PubMed.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638 CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS.
- L. M. Dai, D. W. Chang, J. B. Baek and W. Lu, Small, 2012, 8, 1130 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- T. H. Kim, J. S. Park, S. K. Chang, S. Choi, J. H. Ryu and H. K. Song, Adv. Energy Mater., 2012, 2, 860 CrossRef CAS.
- Y. Q. Wang, L. Gu, Y. G. Guo, H. Li, X. Q. He, S. Tsukimoto, Y. Ikuhara and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 7874 CrossRef CAS PubMed.
- G. Q. Zhang, L. Yu, H. Bin Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609 CrossRef CAS PubMed.
- B. Liu, J. Zhang, X. Wang, G. Chen, D. Chen, C. Zhou and G. Shen, Nano Lett., 2012, 12, 3005 CrossRef CAS PubMed.
- Y. Sharma, N. Sharma, G. V. S. Rao and B. V. R. Chowdari, Bull. Mater. Sci., 2009, 32, 295 CrossRef CAS PubMed.
- Y. Ding, Y. Yang and H. Shao, J. Power Sources, 2013, 244, 610 CrossRef CAS PubMed.
- Q. Q. Xiong, J. P. Tu, S. J. Shi, X. Y. Liu, X. L. Wang and C. D. Gu, J. Power Sources, 2014, 256, 153 CrossRef CAS PubMed.
- Y. Ding, Y. Yang and H. Shao, Electrochim. Acta, 2011, 56, 9433 CrossRef CAS PubMed.
- P. F. Teh, Y. Sharma, S. S. Pramana and M. Srinivasan, J. Mater. Chem., 2011, 21, 14999 RSC.
- F. M. Courtel, H. Duncan, Y. Abu-Lebdeh and I. J. Davidson, J. Mater. Chem., 2011, 21, 10206 RSC.
- Y. Sharma, N. Sharma, G. V. S. Rao and B. V. R. Chowdari, Electrochim. Acta, 2008, 53, 2380 CrossRef CAS PubMed.
- X. Guo, X. Lu, X. Fang, Y. Mao, Z. Wang, L. Chen, X. Xu, H. Yang and Y. Liu, Electrochem. Commun., 2010, 12, 847 CrossRef CAS PubMed.
- J. M. Won, S. H. Choi, Y. J. Hong, Y. N. Ko and Y. C. Kang, Sci. Rep., 2014, 4, 5857 CrossRef PubMed.
- X. Chen, B. Cheng, H. Xu, J. Yang and Y. Qian, Chem. Lett., 2012, 41, 639 CrossRef CAS.
- L. Lian, L. Hou, L. Zhou, L. Wang and C. Yuan, RSC Adv., 2014, 4, 49212 RSC.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622 CrossRef CAS PubMed.
- D. Zhao, Y. Xiao, X. Wang, Q. Gao and M. Cao, Nano Energy, 2014, 7, 124 CrossRef CAS PubMed.
- A. S. Hameed, H. Bahiraei, M. V. Reddy, M. Z. Shoushtari, J. J. Vittal, C. K. Ong and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2014, 6, 10744 CAS.
- X. Tang, X. Hou, L. Yao, S. Hu, X. Liu and L. Xiang, Mater. Res. Bull., 2014, 57, 127 CrossRef CAS PubMed.
- Y. Deng, Q. Zhang, S. Tang, L. Zhang, S. Deng, Z. Shi and G. Chen, Chem. Commun., 2011, 47, 6828 RSC.
- J. Sui, C. Zhang, D. Hong, J. Li, Q. Cheng, Z. Li and W. Cai, J. Mater. Chem., 2012, 22, 13674 RSC.
- D. Bresser, E. Paillard, R. Kloepsch, S. Krueger, M. Fiedler, R. Schmitz, D. Baither, M. Winter and S. Passerini, Adv. Energy Mater., 2013, 3, 513 CrossRef CAS.
- A. Varzi, D. Bresser, J. von Zamory, F. Mueller and S. Passerini, Adv. Energy Mater., 2014, 4, 1400054 Search PubMed.
- B. Luo, S. M. Liu and L. J. Zhi, Small, 2012, 8, 630 CrossRef CAS PubMed.
- J. Kong, Z. Liu, Z. Yang, H. R. Tan, S. Xiong, S. Y. Wong, X. Li and X. Lu, Nanoscale, 2012, 4, 525 RSC.
- J. Kong, W. A. Yee, Y. Wei, L. Yang, J. M. Ang, S. L. Phua, S. Y. Wong, R. Zhou, Y. Dong, X. Li and X. Lu, Nanoscale, 2013, 5, 2967 RSC.
- J. E. Lee, S.-H. Yu, D. J. Lee, D.-C. Lee, S. I. Han, Y.-E. Sung and T. Hyeon, Energy Environ. Sci., 2012, 5, 9528 CAS.
- C. Zhao, J. Kong, X. Yao, X. Tang, Y. Dong, S. L. Phua and X. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 6392 CAS.
- Z. Zhu, S. Wang, J. Du, Q. Jin, T. Zhang, F. Cheng and J. Chen, Nano Lett., 2013, 14, 153 CrossRef PubMed.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394 CrossRef CAS PubMed.
- J. Kong, C. Zhao, Y. Wei, S. L. Phua, Y. Dong and X. Lu, J. Mater. Chem. A, 2014, 2, 15191 CAS.
- J. Kong, W. A. Yee, L. Yang, Y. Wei, S. L. Phua, H. G. Ong, J. M. Ang, X. Li and X. Lu, Chem. Commun., 2012, 48, 10316 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed.
- C. L. Pai, M. C. Boyce and G. C. Rutledge, Macromolecules, 2009, 42, 2102 CrossRef CAS.
- J. Joo, B. Y. Chow, M. Prakash, E. S. Boyden and J. M. Jacobson, Nat. Mater., 2011, 10, 596 CrossRef CAS PubMed.
- Z. Xing, Z. Ju, J. Yang, H. Xu and Y. Qian, Nano Res., 2012, 5, 477 CrossRef CAS.
- H. Xia, Y. Qian, Y. Fu and X. Wang, Solid State Sci., 2013, 17, 67 CrossRef CAS PubMed.
- J. Xie, W. Song, G. Cao, T. Zhu, X. Zhao and S. Zhang, RSC Adv., 2014, 4, 7703 RSC.
- L. Yao, X. Hou, S. Hua, X. Tang, X. Liu and Q. Ru, J. Alloys Compd., 2014, 585, 398 CrossRef CAS PubMed.
- Y. He, L. Huang, J. S. Cai, X. M. Zheng and S. G. Sun, Electrochim. Acta, 2010, 55, 1140 CrossRef CAS PubMed.
- Y. Hwa, J. H. Sung, B. Wang, C.-M. Park and H. J. Sohn, J. Mater. Chem., 2012, 22, 12767 RSC.
- J. R. Dahn, T. Zheng, Y. H. Liu and J. S. Xue, Science, 1995, 270, 590 CAS.
- R. Mukherjee, A. V. Thomas, D. Datta, E. Singh, J. Li, O. Eksik, V. B. Shenoy and N. Koratkar, Nat. Commun., 2014, 5, 3710 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA curves of PS porous nanofibers (PNFs) and PDA-coated PS PNFs from 2-times coating, TGA curve of the introduction of ZnFe2O4 nanoneedles into PNFs-2, the morphology of the Zn complexes/Fe(OH)3 nanoneedles on the C-PDA porous nanofibers before annealing, galvanostatic discharge–charge profiles (1st, 2nd and 3rd cycle) of the C-PDA porous nanofibers (PNFs-6) as a LIB anode. See DOI: 10.1039/c4ra16460a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 







