
A simple and versatile approach to self-healing polymers and electrically conductive composites†
Abstract
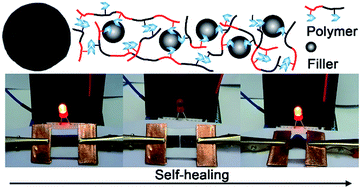
In this study, a simple and versatile approach to self-healing polymers and electrically conductive composites is reported. A series of self-healing polymers are readily synthesized by radical copolymerization of two acrylate monomers bearing a hydrogen-bonding motif. Subsequent blending with nanofillers leads to self-healing electrically conductive composites. Their glass transition temperature, the mechanical and electrical properties, and the self-healing capability can be readily tuned by the composition of the polymer as well as the filler fraction. The composite exhibits mechanical force sensing capabilities and high efficiency of both mechanical and electrical self-healing properties. Our design starts from simple chemistry and inexpensive materials, and may offer a new route towards economic self-healing electronic/electrical devices.
 Please wait while we load your content...
Please wait while we load your content...

