
Diverse zinc(ii) coordination assemblies built on divergent 4,2′:6′,4′′-terpyridine derivatives: syntheses, structures and catalytic properties†
Abstract
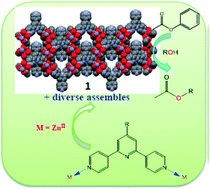
A series of metal–organic architectures (compounds 1–5) based on zinc salts and four 4′-substituted 4,2′:6′,4′′-tpys including two new ligands have been synthesized and structurally characterized. The ligands L1–L4 with various 4′-substituents on 4,2′:6′,4′′-tpy react with Zn(OAc)2·2H2O to yield assemblies 1–4 containing either 1-D polymeric chains (1–3) or a discrete dinuclear complex (4). X-ray structural analysis revealed that although similar 1-D polymeric chains were observed in both 1 and 3, the 3-D packing modes were essentially different. The chains in 1 were stacked to form a network structure with microporous channels that were not present in 3. In contrast, 4 was a discrete dinuclear complex containing a paddle-wheel {Zn2(μ-OAc)4} motif, although another minor component possibly coexists in the bulk sample as revealed by the PXRD studies. Crystals of 5 were prepared from the reaction between L4 and ZnI2 and its X-ray structure revealed a 1-D polymeric chain with a wave-like structure. Phase-pure compounds 1–3 and 5 were tested for the catalytic transesterification of phenyl acetate with alcohols, and the results indicated that 1 was the most active catalyst for this reaction, affording the new ester product in 95% yield at 50 °C under neat conditions, while other catalysts also catalysed the reaction with modest yields. Several different alcohols were examined as substrates for 1-catalysed transesterification and it was found that the size of substrates has important influence on the catalytic efficiency. In addition, amine additives were found to remarkably promote the catalytic efficiency of the less active catalyst 3. The structure–catalytic activity relationship was discussed in detail based on the catalytic data obtained.
 Please wait while we load your content...
Please wait while we load your content...

