DOI:
10.1039/C4RA16245E
(Paper)
RSC Adv., 2015,
5, 15880-15885
Visible-light-driven photocatalytic H2 evolution from aqueous suspensions of perylene diimide dye-sensitized Pt/TiO2 catalysts
Received
12th December 2014
, Accepted 28th January 2015
First published on 28th January 2015
Abstract
TiO2 is among the most important semiconductor photocatalysts for solar light-driven H2 production from photocatalytic water reduction. Commercialized mixed-phase Degussa P25 nanoparticles have been demonstrated as the most popular TiO2. However, its superiority cannot be exerted in the visible-light region. In this work, molecular aggregates of perylene tetracarboxylic diimide (PTCDI, a kind of air-stable n-type organic semiconductor bearing an electron-rich phenylamino moiety and a dodecyl side-chain) were employed as a sensitizer to combine with Pt/TiO2 nanoparticles via solution processing. The PTCDI/Pt/TiO2 composites thus fabricated exhibit a broad visible-light response and excellent photochemical stability. The initial intramolecular charge transfer of PTCDI and its energy level being well matched to TiO2, ensure effective charge transfer among the PTCDI/Pt/TiO2 composite. When 25 mg of the composite powders are suspended in aqueous solution (100 mL) containing 10 mL of triethanolamine as a sacrificial electron donor, stable H2 evolution with an activity of ∼0.075 μmol h−1 is achieved under visible-light (λ ≥ 420 nm) irradiation. Both intramolecular electron transfer from the electron rich 4-dimethylaminobenzyl group to the perylene core of PTCDI and the intermolecular charge transfer from PTCDI to TiO2 are key to the effective visible-light photoactivity.
Introduction
Photocatalytic water reduction over semiconductor nanoparticles utilizing solar light has been recognized as one of the most promising approaches to produce H2, an ideal clean and renewable energy source with high energy capacity.1–5 The work was initiated in the 1970s with an inorganic semiconducting photocatalyst, titanium dioxide (TiO2).6 In contrast to many other inorganic semiconductors (e.g., metal oxides and sulfides),2 graphitic carbon nitride,7 crystalline polyimides,8 and carbon materials,9–12 which have attracted intense interest in subsequent years, TiO2 benefits from being cost-saving, effective and photochemically stable.13 Very promising, high-activity, mixed-phase TiO2 nanoparticles have been brought into the market under the brand of Degussa P25. Despite its appealing features, low photoconversion efficiency still limits its use in practical applications.14 The main obstacles are attributed to the following two aspects: its large bandgap (Eg = ∼3.2 eV), thus it can only be excited by ultraviolet light (only covering 4% of the solar spectra), and the rapid bulk and surface recombination of photoinduced electrons and holes.2,5 Consequently, increasing research efforts have now been put into eliminating these drawbacks.15 Normally, loading of Pt nanoparticles as cocatalyst on the surface of TiO2 nanoparticles and adding sacrificial electron donors (e.g., triethanolamine and methanol) to the aqueous solutions are often necessary for photocatalytic H2 production from water by inhibiting the fast charge recombination and the back reaction of H2 and O2 to water.6 Additionally, adsorption of dye molecules on the surface of TiO2 nanoparticles has been proven as one of the most facile approaches to utilize visible-light (∼43% of the solar spectra).14,16 However, most reported photosensitizing dyes suffer from disadvantages such as expensive, thermally unstable, photobleaching, limited visible-light response, or difficult functionalization.6,14,17–20 Therefore, it is highly desirable to develop novel dyes in this aspect.
Perylene diimides, a series of well-known n-type organic semiconductor molecules, exhibit unique photochemical and thermal stability under long-term light irradiation, and strong visible-light response.21–23 In contrast to the extremely attractive applications of perylene diimide-based semiconductor systems for fabricating organic optoelectronic devices, such as organic photovoltaic cells, organic field effect transistors, organic light-emitting diodes, dye lasers, sensors, and so on,21–24 very limited appealing results on photocatalysis field have been reported in the last decades. Recently, significant efforts have focused on the applicability of a series of hybrid photocatalyst composites, TiO2 coated nanofibers of perylene diimides, for visible-light-driven H2 generation from photocatalytic water reduction.25 Although the fabrication of TiO2 coatings from organic titanium complex precursors via in situ hydrolytic processing was facile, the amorphous feature of TiO2 limits their photoactivity compared to well crystallized Degussa P25.14,15 Moreover, taking advantage of fine structural modification, perylene tetracarboxylic diimide (PTCDI; Scheme 1) modified with electron-donating 4-dimethylaminobenzyl group and inert dodecyl chain at the imide-position of the electron-deficient π-conjugated core has displayed much more effective intramolecular charge separation than 4-dimethylaminophenyl group modification, and exhibits excellent charge transport along one-dimensional nanofibers (self-assembled from a bisolvent interfacial transfer process) under visible-light.25–27 According to electrochemical cyclic voltammetry (CV) measurement, PTCDI has energy levels (vs. vacuum level) at the positions of −6.3 eV and −3.7 eV for the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), respectively.26 This ensures its visible-light absorption corresponding to a bandgap of 2.6 eV and its matched energy levels to TiO2 (conduction band (CB): −4.2 eV; valance band (VB): −7.4 eV).6 These features make PTCDI as a candidate for visible-light harvesting sensitizer in commercial TiO2-based photocatalytic water reduction system.
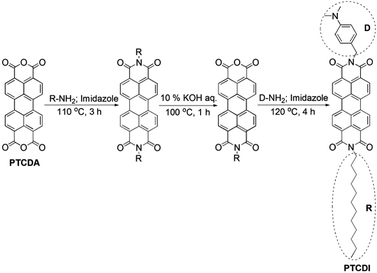 |
| | Scheme 1 Synthesis route and chemical structure of PTCDI molecule. | |
In this work, to develop a facile and efficient solar water reduction H2 generation system, a novel visible-light-driven composite photocatalyst employing commercial TiO2 (P25; pre-deposited with cocatalyst Pt) as the main body and PTCDI assembles as the photosensitizer were fabricated. Asymmetric PTCDI was synthesized following a typical Langals' procedure reported before (Scheme 1).25,26 After photo-deposition of cocatalyst Pt on TiO2, a bisolvent-exchange triggered aggregation strategy was used to prepare PTCDI/Pt/TiO2 composite. The weight ratio of PTCDI to Pt/TiO2 was adjusted to tune the photocatalytic activity. Following that, the composite particles were suspended in aqueous solution containing triethanolamine (sacrificial electron donor) to sustain the photocatalytic reaction under visible-light illumination. In comparison, the photocatalytic activity of Pt/TiO2 and another composite fabricated by mixing PTCDI solution and Pt/TiO2 together were also explored to evaluate the photocatalytic activity of PTCDI/Pt/TiO2 composites.
Experimental section
Chemicals and materials
TiO2 nanoparticles, P25 (Fig. 1a), was supplied by Degussa, Germany. Perylene 3,4,9,10-tetracarboxylic dianhydride (PTCDA; Scheme 1), 4-(dimethylamino)benzylamine, dodecylamine and hexachloroplatinic acid hexahydrate (H2PtCl6·6H2O) were purchased from J&K Acros. All other chemicals were obtained from Chemical Reagent Co. Ltd. (Shanghai, China) in analytical grade and used without any purification.
 |
| | Fig. 1 Photo and SEM images of (a) TiO2, (b) Pt/TiO2, and PTCDI/Pt/TiO2 composites containing PTCDI with different weight ratios of (c) 0.5%, (d) 1% and (e) 2%. | |
Preparation of PTCDI/Pt/TiO2 composites
10 mL of PTCDI solution in CHCl3 (12.5, 25 or 50 mg L−1) was mixed with Pt/TiO2 powders (25 mg; weight ratio of Pt to TiO2 is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200; Fig. 1b) in a 50 mL round-bottom flask. The mixture was stirred at 25 °C for 6 h in the dark, and then for another 2 h after the dropwise injection of 40 mL of CH3OH. The precipitate was collected by vacuum suction filtration using 0.45 μm membrane filter and then dried at 80 °C for 6 h to give composites, denoting as 0.5%, 1% or 2% PTCDI/Pt/TiO2 (Fig. 1c–e). For comparison, 5 mL of CHCl3 solution of PDI (1% of the weight of Pt/P25) was mixed with grinded Pt/TiO2 fine powder (0.025 g) in a mortar. The mixture was grinded for 5 min, and then the powder was collected via evaporation for comparative photocatalytic experiment.
:200; Fig. 1b) in a 50 mL round-bottom flask. The mixture was stirred at 25 °C for 6 h in the dark, and then for another 2 h after the dropwise injection of 40 mL of CH3OH. The precipitate was collected by vacuum suction filtration using 0.45 μm membrane filter and then dried at 80 °C for 6 h to give composites, denoting as 0.5%, 1% or 2% PTCDI/Pt/TiO2 (Fig. 1c–e). For comparison, 5 mL of CHCl3 solution of PDI (1% of the weight of Pt/P25) was mixed with grinded Pt/TiO2 fine powder (0.025 g) in a mortar. The mixture was grinded for 5 min, and then the powder was collected via evaporation for comparative photocatalytic experiment.
Photocatalytic tests
H2 generation experiments were carried out in a top-irradiation type reactor connected to a closed gas circulation and evacuation system (Labsolar-H2 photocatalytic water-splitting H2 production system, Bofeilaii, Beijing). Generally, 25 mg of sample photocatalyst powder was suspended using a magnetic stirrer in 100 mL aqueous solution containing triethanolamine (10% by volume) as sacrificial electron donor. Before photo-irradiation, the system was evacuated to remove oxygen, and the reaction temperature was maintained at 25 °C by a flow of water during the entire experiment. A 400 W Xenon lamp (CEL-S500, Beijing AULTT) attaching with a 420 nm cutoff optical filter, which blocked the light with wavelengths of below 420 nm, was used as the light source. The intensity of the incident light (≥420 nm) was measured to be ∼150 mW cm−2 with a optical power meter (NEWPORT 1916-C). The effective irradiation surface area is 19.6 cm2. The amount of evolved H2 was detected and analyzed by an inline gas chromatography (GC-system 7890A; Agilent Technologies) with N2 as carrier gas. The photocatalytic test under monochromatic light irradiation was also conducted under the same photocatalytic reaction condition by using relevant band-pass filter (with wavelength of 365, 380, 420, 450, 500, 550 and 610 nm possessing full width at half maxima of 15 nm). The apparent quantum efficiency (AQE) of H2 evolution was calculated over 0.25 g of optimal PTCDI/Pt/TiO2 catalyst according to the typical formula, AQE = [(number of reacted electrons)/(number of incident photons)] × 100% = [2 × (number of evolved H2 molecules)/(number of incident photons)] × 100% = [(2·n·t·N)/(E·A·t·λ/h/c)] × 100%, where n is the amount of H2 evolution under the light with the wavelength λ, t is the illumination time, N is Avogadro constant, E is the irradiated light intensity, A is the effective irradiation surface area, h is the Planck constant, and c is the speed of light.
Characterization
Optical properties were analyzed with a UV-vis spectrophotometer (SHIMADZU UV-1800). UV-vis Diffused Reflectance Spectra (DRS) of the samples were recorded at room temperature on an EVOLUTION 220 UV-visible spectrophotometer equipped with an integrating sphere assembly. Scanning electron microscopy (SEM) images were obtained on a ZEISS SUPRA55VP microscope.
Results and discussion
Materials and characterization
Commercial TiO2 nanoparticles (Fig. 1a) were loaded with traces of cocatalyst Pt via a simple in situ photo-deposition procedure, resulting in a color change varying from white to grayish (Fig. 1b) and a red-shift of the absorption band edge to some extent according to the Kubelka–Munk function but still lies within 400 nm (Fig. 2). Thus, to develop a functional visible-light-driven photocatalytic system, PTCDI is implemented to photosensitize TiO2 forming a kind of composite PTCDI/Pt/TiO2 (Fig. 1c–e). It is known that the organization of PTCDI molecules into aggregates can provide continuous pathway for charge transport.24 Therefore, a solution processing approach derived from the reported self-assembly strategies of perylene diimides is initially introduced to prepare PTCDI/Pt/TiO2 composite as stated in the Experimental section.24,26 For balancing the aggregation behavior and solubility, PTCDI quite benefits to its asymmetric structure modified with 4-dimethylaminobenzyl group and long dodecyl chain at N,N′-positions. It is facilely soluble in CHCl3 while not in CH3OH. Stacking of PTCDI molecules surround Pt/TiO2 nanoparticles (in suspending state) occurs as their solubility decreases when injecting a fourfold volume of CH3OH into their CHCl3 solution, which is mainly driven by their strong intrinsic co-facial π–π stacking of the flat aromatic core (stacking along the long axis) in conjunction with the association between the side chains (aggregating along the short axis).24,25 This method bears the advantages of low-cost, low power consumption, easy scaling, and simplicity. The robust visible-light harvesting property of PTCDI and the aggregation process can be followed by UV-vis absorption spectra in Fig. 3, where a novel absorption peak among 550–600 nm appears accompanied by the transformation of monomeric molecules to aggregates.24 As can be seen from the DRS measurements (Fig. 2), the absorption range of Pt/TiO2 (below 400 nm) extends to the visible range (400–700 nm) after combining with PTCDI aggregates, which makes the PTCDI/Pt/TiO2 composite as a good candidate for visible-light-driven photocatalyst. In view of the weight ratio difference between PTCDI and Pt/TiO2, more PTCDI components in the composites lead to stronger visible-light absorption (Fig. 2) as evidenced by their much darker color (Fig. 1). However, it should be pointed that the contact of PTCDI with Pt/TiO2 is still not so strong as chemically bonding (e.g., via covalent bond),14 thus, the effect of PTCDI on the absorption edge of Pt/TiO2 is not so obvious. As a result, in the composite, PTCDI acted only as the photosensitizer to utilize the photos of light above 420 nm. Further, the photocatalytic activity may be tuned due to the strong electron-donor feature of 4-dimethylaminobenzyl group to electron-deficient PTCDI unit, constructing fast intramolecular charge transfer among donor–acceptor type PTCDI.25,26 Under visible-light illumination, photo-excitation of PTCDI promotes electron transition from the HOMO to the LUMO, while TiO2 works as electron transfer relay that accepts electron from PTCDI and transfers it to Pt, which provides active sites for reduction of water to H2. However, it is speculated that more PTCDI didn't give rise to better photocatalytic activity because the contact area between PTCDI and Pt/TiO2 is confined and excess amount of PTCDI aggregates may deteriorate the dispersity of the composite in aqueous solution. This is supported by SEM measurements (Fig. 1) where in the presence of 2% PTCDI, the composite shows a kind of clump-like morphology.
 |
| | Fig. 2 DRS Kubelka–Munk and absorption (inset) spectra of TiO2, Pt/TiO2, and PTCDI/Pt/TiO2 with different weight ratio of PTCDI to Pt/TiO2. | |
 |
| | Fig. 3 UV-vis absorption spectra of PTCDI in CHCl3 solutions with different concentrations, and recorded by adding 50 vol% CH3OH (inset). | |
Photocatalytic H2 production
As shown in Fig. 4, H2 evolution from PTCDI/Pt/TiO2 composite system is stable and proceeds at a rate up to 0.075 μmol h−1 in contrast to nearly inactive Pt/TiO2 under visible-light (λ ≥ 420 nm) illumination. The weight ratio of PTCDI component to Pt/TiO2 was optimized at 1% to balance the surface coverage and dispensability, resulting into the highest performance followed by 2% and 0.5%. It has been found that excess (2%) aggregates of PTCDI on the surface of Pt/TiO2 nanoparticles can obstruct their active cores for H2 generation and limited the dispersity of the constructed composite in aqueous solution, while fewer amounts of PTCDI aggregates (0.5%) may lead to uncovered Pt/TiO2. The ratio of 1% is the appropriate choice to maintain good dispensability and avoid clumping of the composite during the construction process. The above statements are supported by the images as shown in Fig. 1. For mixed PTCDI/Pt/TiO2 system, much poor photocatalytic efficiency is achieved probably due to the unsatisfied interfacial action between PTCDI and Pt/TiO2. In other words, the weak contract between them results in inferior interfacial charge transfer.25 Conversely, the good photocatalytic performance of the sample via solution processing method is largely due to the bisolvent triggered aggregation of PTCDI molecules. The PTCDI assemblies thus fabricated may play a role as glue to sticking Pt/TiO2 nanoparticles together via forces such as hydrogen bonding and π–π stacking. This structure in combination with π–π stacking morphology of PTCDI enables efficient interfacial charge transfer and separation to enhance photocatalytic activity.25 In addition, it has been demonstrated that nanofiber composite of Pt/PTCDI without TiO2 shows poor activity (ca. 0.025 μmol h−1) under the same conditions, which is attributed to the competition between intrinsic inefficient photoactivity of organic dyes without a semiconductor matrix to act as an efficient charge separator and the well-defined 1D nanofiber morphology for enhanced charge transfer.25 Therefore, a synergetic role is involved in the system of PTCDI (photosensitizer) to Pt (photoreduction reaction core) deposited TiO2 (photocatalyst). This conclusion is also supported by the photocatalytic test under monochromatic light illumination. In general, a wavelength dependence feature of the incident light on the rate of H2 evolution over optimal 1% PTCDI/Pt/TiO2 catalyst has been demonstrated. As seen in Fig. 5, the AQE data are quite consistent with the H2 evolution rate in accordance with the absorption spectrum of 1% PTCDI/Pt/TiO2. Thus, it can be speculated that under the irradiation of light with the wavelength above 420 nm, it is PTCDI molecules who act as the light-absorber. Especially, an AQE as high as 0.47‰ is achieved under the irradiation of 550 nm monochromatic light.
 |
| | Fig. 4 The time courses of H2 evolution from water over Pt/TiO2 and various PTCDI/Pt/TiO2 composites with assistance of visible-light irradiation. | |
 |
| | Fig. 5 Absorption spectrum and the photocatalytic activity of 1% PTCDI/Pt/TiO2 for H2 evolution under monowavelength light irradiation. The inset figure shows the dependence of the AQE on the wavelength of monochromatic visible-light. | |
Further, the stability of the photocatalytic activity of 1% PTCDI/Pt/TiO2 for H2 evolution under visible-light illumination was observed in Fig. 6. As both PTCDI and TiO2 are stable under light irradiation in aqueous triethanolamine solution, the photocatalytic reaction results in a stable H2 evolution rate, although there is still a decrease by ∼10% during 36 h probably due to the interface separation between PTCDI and TiO2 during stirring process. Furthermore, PTCDI molecules are stable enough to inhibit photobleaching. They can be regenerated by dissolving the reacted composite powders into CHCl3.
 |
| | Fig. 6 Stable H2 evolution from water over 1% PTCDI/Pt/TiO2. The reaction was continued for 36 h, and reset every 12 h. | |
Mechanism speculation
To explain the visible-light-driven photocatalytic H2 generation performance of PTCDI/Pt/TiO2 from aqueous solution in the presence of sacrificial reagent triethanolamine, both the intramolecular electron transfer from the electron rich 4-dimethylaminobenzyl group (donor part) to the perylene core (acceptor part) of PTCDI and the intermolecular charge transfer from PTCDI to TiO2 due to the energy level difference are taken into consideration. As illustrated in Scheme 2, under visible-light illumination, only PTCDI can be excited by absorbing photons with the light wavelengths above 420 nm. On one hand, an ultra-fast intramolecular charge transfer process occurs, that is, after the excitation of PTCDI, the electrons can be transferred from the 4-(dimethylamino)benzyl moiety to the HOMO orbital of the photoexcited PTCDI core to quench the photogenerated holes. Due to the presence of large amount of sacrificial reagent triethanolamine (electron donor), the oxidized (dimethylamino)benzyl is easily reduced to its ground state. These processes highly inhibit the recombination of photo-generated electron–hole pairs among PTCDI. Consequently, residual excited photogenerated electrons are left on the LUMO of PTCDI. On the other hand, due to the energy level fall from the LUMO of PTCDI to the CB of TiO2, the rich excited electrons of PTCDI move to the CB of TiO2 via interface charge transfer. These electrons are captured by the loaded Pt nanoparticles (electron traps) on TiO2, thus promoting the reduction of water to H2 by providing active sites and suppressing the back charge recombination.6 The electrons are consumed by water to proceed the reduction reaction process for H2 production. Such TiO2 mediated electron transfer from PTCDI to Pt helps suppress the electron–hole recombination of PTCDI. Further, the π–π stacking morphology of PTCDI in the composite is also good for its photocatalytic performance by providing large electron mobilized terrace for PTCDI and TiO2 nanoparticles, which can enhance the intra-/inter-molecular charge transfer among the composite.25 As a result, PTCDI can be identified as an efficient visible-light-responsive sensitizer for TiO2.
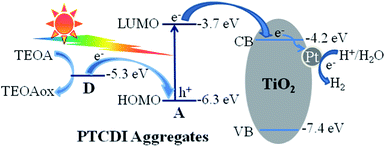 |
| | Scheme 2 Proposed mechanism for photocatalytic H2 evolution over PTCDI/Pt/TiO2 under visible-light (λ ≥ 420 nm). D: 4-dimethylaminobenzyl moiety of PTCDI; A: perylene core of PTCDI; TEOA: triethanolamine. Energy values (vs. vacuum level) are taken from ref. 25 and 26. | |
Conclusions
To sum up, PTCDI/Pt/TiO2 composite is demonstrated as an efficient photocatalyst for visible-light-driven H2 evolution from photocatalytic water reduction. Two key design criteria for the application of PTCDI are involved: one is the electron donor–acceptor feature of PTCDI resulting from the presence of electron-rich 4-dimethylaminobenzyl moiety; and the other is bisolvent-induced π–π stacking morphology with the assistance of dodecyl modification. Although the present photocatalytic activity of this composite is still unsatisfied, perylene diimide sensitized commercial TiO2 nanoparticles hold great potential for practical photocatalytic water reduction processes due to their excellent solar light harvesting ability, good photochemical and thermal stability, and flexible structural design of perylene diimides.27 Moreover, the simple solution-induced processing method demonstrates a new strategy to develop economical organic–inorganic composite photocatalysts. More efforts are needed in tuning the structures of perylene diimide molecules (e.g., introducing functional side-groups to improve their binding ability with TiO2) and tunable composite fabrication.28
Acknowledgements
Financial support by the National Natural Science Foundation of China (Grant no. 21173261), the “One Hundred Talents” program of Chinese Academy of Sciences (Grant no. 1029471301), and the “Cross Cooperation Program for Creative Research Teams” of Chinese Academy of Sciences (Grant no. Y251821601) is gratefully acknowledged.
Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS PubMed.
- S. W. Cao and J. G. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef CAS.
- S. Chu, Y. Wang, C. C. Wang, J. C. Yang and Z. G. Zou, Int. J. Hydrogen Energy, 2013, 38, 10768–10772 CrossRef CAS PubMed.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- Q. J. Xiang and J. G. Yu, J. Phys. Chem. Lett., 2013, 4, 753–759 CrossRef CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- N. Zhang, Y. H. Zhang and Y. J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- W. J. Youngblood, S. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966–1973 CrossRef CAS PubMed.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- J. Schneider, M. atsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- S. Caramori, V. Cristino, L. Meda, R. Argazzi and C. A. Bignozzi, Top. Curr. Chem., 2011, 303, 39–94 CrossRef CAS.
- J. Zhang, P. W. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2007, 129, 7726–7727 CrossRef CAS PubMed.
- W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- X. H. Zhang, L. J. Yu, C. S. Zhuang, T. Y. Peng, R. J. Li and X. G. Li, RSC Adv., 2013, 3, 14363–14370 RSC.
- S. G. Li, Y. W. Guo, L. Zhang, J. Wang, Y. Li, Y. Li and B. X. Wang, J. Power Sources, 2014, 252, 21–27 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS PubMed.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1606 CrossRef CAS PubMed.
- S. Chen, D. L. Jacobs, J. K. Xu, Y. X. Li, C. Y. Wang and L. Zang, RSC Adv., 2014, 4, 48486–48491 RSC.
- Y. Che, X. M. Yang, G. L. Liu, C. Yu, H. W. Ji, J. M. Zuo, J. C. Zhao and L. Zang, J. Am. Chem. Soc., 2010, 132, 5743–5750 CrossRef CAS PubMed.
- L. E. Shoer, S. W. Eaton, E. A. Margulies and M. R. Wasielewski, J. Phys. Chem. B, 2014 DOI:10.1021/jp511624s.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 







