
Multiple deuteration of alkanes synergistically-catalyzed by platinum and rhodium on carbon as a mixed catalytic system†
Abstract
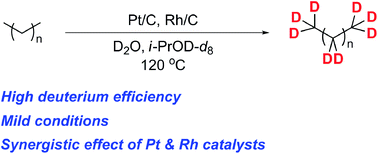
We have accomplished an efficient and mild multiple deuteration method for alkanes catalyzed by the combined use of heterogeneous platinum on carbon (Pt/C) and rhodium on carbon (Rh/C) catalysts in i-PrOD-d8 and D2O as a mixed solvent. The present multi-deuteration could be initiated by the transition metal-catalyzed dedeuteration of i-PrOD-d8 to produce D2 and the subsequent C–H bond activation of alkanes catalyzed by the Pt/C and/or Rh/C–D2 complex. This method could be applied to the deuteration of wide variety of linear, branched and cyclic alkanes as useful deuterated materials under mild conditions.
 Please wait while we load your content...
Please wait while we load your content...

