
A comprehensive spectroscopic investigation of α-(2-naphthyl)-N-methylnitrone: a computational study on photochemical nitrone–oxaziridine conversion and thermal E–Z isomerization processes†
Praveen Saini and
Anjan Chattopadhyay*
Department of Chemistry, Birla Institute of Technology and Science (BITS), Pilani – K.K. Birla Goa Campus, Goa, 403 726, India. E-mail: anjan@goa.bits-pilani.ac.in; anjan_chattopadhyay@yahoo.com; Fax: +91 832 2557033; Tel: +91 832 2580319
First published on 18th February 2015
Abstract
This comprehensive spectroscopic analysis of α-(2-naphthyl)-N-methylnitrone has proposed its photochemical oxaziridine formation and thermal E–Z isomerization mechanisms. The activation energy for the conversion of the unstable non-planar E isomer to the stable planar Z-isomer is found to be 23.7 kcal mol−1 at the CASSCF/6-31G* level of calculation. A transition state with a negative frequency of 350 cm−1 is likely to be responsible for this process. Both CASSCF and ONIOM-based studies have revealed that the nitrone–oxaziridine photochemical conversion involves non-radiative decay channels which include biradicaloid conical intersection (CI) points through Hula-twist and terminal one-bond-flip motions, situated at 35–40 kcal mol−1 below the first excited singlet state (S1). Following the directions of their gradient-difference vectors, the optimized oxaziridine geometries are obtained. The nature of the low-lying singlet–singlet transitions of these α-naphthyl N-methylnitrones is found to be similar to that of the conjugated non-polar polyenes, and differs appreciably from our previously studied long-chain conjugated nitrone systems. The fluorescent S1 state with a radiative lifetime of nanosecond order is populated by the weak upward S0–S1 transition (transition moment: 0.3 Debye) and through the decay of the S2 state, which eventually gets involved in the S0/S1 conical intersections.
1. Introduction
Nitrones are well-known for their stereoselective formation of synthetically useful isoxazolidines by 1, 3-dipolar cycloaddition with alkenes.1–3 In the last few years, nitrones have also drawn special attention due to their spin-trapping properties.4–6 Nitrone spin traps react with transient free radicals to form more persistent paramagnetic species, known as spin-adducts and these are used to detect transient radicals in electron paramagnetic resonance (EPR) spectroscopy. They are known to have neuroprotective and antiaging properties,7,8 and used in the treatment of inflammatory and degenerative age-related diseases, such as Alzheimer's disease.9 There are numerous examples where nitrones act as important pharmacological agents in several other diseases,10–16 too. One such example is the chemopreventive conjugated long-chain N-alkyl retinylnitrones,17 those were synthesized and analyzed almost thirty years back. Recently our group has carried out computational studies18 on the model compounds of these nitrones and proposed their photochemical oxaziridine conversion mechanism. This process is found to include a non-radiative decay channel through the lowest-energy biradicaloid conical intersection geometry, leading to oxaziridine as their primary photoproduct.The photochemical conversion of nitrone–oxaziridine process has been experimentally attempted by several groups in the last few decades.19,20 The photoirradiation studies on nitrones can be primarily summarized into 2 important observations; the cis–trans isomerization reactions of nitrones occur thermally or through triplet excited states in presence of photosensitizers,21,22 while their normal photo-excitation involves singlet excited states which subsequently forms oxaziridines and other photo-products, such as amides. The other noticeable feature of the nitrone photo-excitation is the varying stability of the photoproduct oxaziridine, which largely depends on the substituents present on nitrogen or α-carbon.19,23 Experimental results have shown that N-alkyl substituents increase the stability of the oxaziridines and this has also been established by our CASSCF-based studies on model N-alkyl retinyl nitrone systems.18 This present work is aimed to reveal the unexplored mechanism of photochemical processes of one such nitrone system which is known to give a stable oxaziridine species on photo-irradiation; the experimentally studied α-(2-naphthyl)-N-methylnitrone was found23 to give stable and isolable oxaziridines, while its N-(p-tolyl) derivative was unable to produce such photoproduct. Photo-irradiation of neutral solution of this nitrone was found to increase the fluorescence intensity with fluctuations which was predicted to be arising due to the formation of efficiently fluorescing oxaziridines.
This study is an attempt to put forward a proper theoretical background of the nitrone–oxaziridine conversion and E–Z isomerization processes of this above-mentioned nitrone system through high-level quantum mechanical investigations. This analysis will include the study of optimized ground state, relaxed excited state, conical intersection points and important transition state geometries. In recent times, computational chemists have been able to develop tools to explore electronically excited state species. Intensive computational studies have revealed the behaviour of the photo-excited organic molecules, and highly systematic investigations are currently possible for the photochemical reaction pathways. In addition to the radiative emission, computational studies involving non-radiative processes have also drawn considerable attention in the last few decades. In a photochemical process, the electronically excited state has a high probability of passing through a region where the excited state crosses the ground state, thereby causing the breakdown of Born–Oppenheimer approximation. Such non-adiabatic crossing, commonly known as conical intersection,24–30 opens up a funnel-type radiationless decay channel, which results in subsequent photoproduct formation.
Our earlier studies on the model compounds of N-alkyl retinyl nitrones have predicted the presence of terminally twisted biradicaloid conical intersection (S0/S1) geometries with a C–N–O kink as the major player behind their photochemical oxaziridine conversion process. This was found to have some similarity with the prefulvenic conical intersection in benzene which appears during its channel-3 decay.30–32 It will be quite interesting to investigate whether our presently targeted nitrone system also involve similar conical intersection topography during the oxaziridine formation step as the retinyl nitrones or not. In addition to this non-radiative channel, analysis of their radiative transition properties will be also an important part of this work, and they are expected to give some equally important features. Their UV-vis peaks will be compared with the experimentally reported values. In addition to these comprehensive studies of the photochemical and photophysical properties of the α-(2-naphthyl)-N-methylnitrone, the probable mechanism of its E–Z isomerization process will be also analyzed in this present work.
2. Computational details
Calculations based on the Complete Active Space Self-Consistent Field (CASSCF) method33,34 and the Our own N-layer Integrated molecular Orbital and Molecular mechanics method35–41 (ONIOM) have been employed for locating the minimum energy geometries, transition states and conical intersection points on the potential energy surfaces (PES) through Gaussian 09 program.42 In addition to these methods, the ground states of both the isomers were also optimized at the Restricted Hatree–Fock (RHF), Density Functional Theory (DFT) and PM3/CI level of calculations. Multiconfiguration-based methods, such as CASSCF33,34,43 and CASMP2 (ref. 44 and 45) are known to be highly accurate quantum mechanical techniques in exploring the excited state topographies of molecules. However, use of large active spaces in these methods may be troublesome in many systems and the computational costs are often too high. It is a fact that reducing an active space size without any prior knowledge of the actual reaction path can certainly bring huge errors in the computational results, but in case we have some prior experimental knowledge of the studied reaction path, certain intuitions may help us to choose an accurate minimal active space, leaving out the less important orbitals (for that particular reaction) from the calculations. Moreover, use of a large active space in such reactions may take it away to some other directions instead of leading to the desired product. Our current calculations are mostly intended to emphasize the importance of the identified active part of the nitrone molecule participating in the experimentally studied nitrone–oxaziridine photochemical conversion process.In our earlier work18 it was concluded that the open-chain conjugated nitrone systems experience a twist at the terminal C–N–O moiety during the oxaziridine formation step without any significant change in the conjugated chain part. Based on the experimental and theoretical studies on the N-methyl retinyl nitrone system, the actual photochemical reaction path of such conjugated nitrone systems leading to oxaziridine is shown in Scheme 1. CASSCF studies had confirmed that the lowest-energy conical intersection geometry related to this terminal CNO twist is responsible for the formation of the oxaziridine ground state species. This study has indicated an involvement of the C–N π bond and a pz orbital on oxygen (holding the negative charge on oxygen) in the oxaziridine formation which results in a C–N σ bond and a possible carbon–oxygen bond (through a transient biradical species), after photo-excitation. Our currently studied α-naphthyl N-methyl nitrone is also likely to follow a similar path, as experimental studies have shown that no change in the naphthyl part was found to happen during the course of oxaziridine formation.
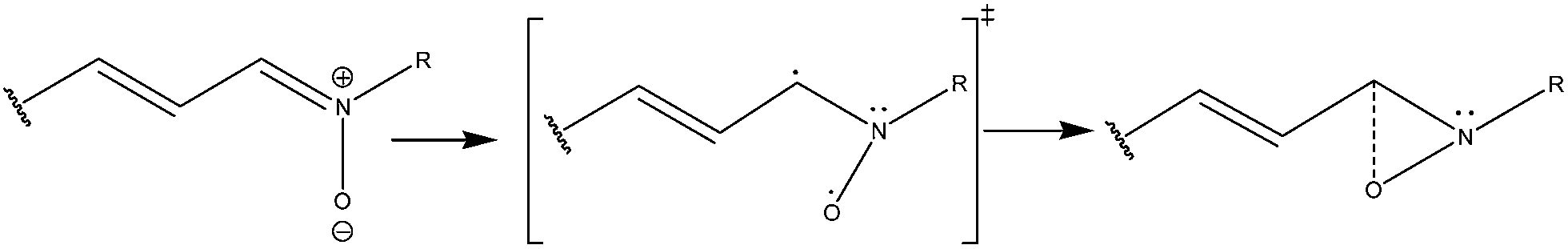 | ||
| Scheme 1 A possible route of nitrone–oxaziridine conversion in case of N-methyl retinylnitrone. | ||
Different regions of a chemical system can be treated with a different level of theory in the hybrid ONIOM method. The basic idea behind this methodology is related to the fact that for a large molecule the major chemical process is usually localized only in a small segment, commonly known as the active site. This zone of our interest where the chemical process is actually happening can be treated with a higher level of quantum mechanical (QM) theory, while the less important part of the system can be treated with a lower-level of QM theory (QM′) or molecular mechanics (MM) theory. Considering the fact that a similar photochemical path as the model retinyl nitrone system is possibly operating in our currently studied nitrone system, it seems quite reasonable if we consider the terminal C–N–O moiety as the active site for the ONIOM calculation. It must be emphasized here that our major goal in this work is to find out the possible route of the experimentally studied oxaziridine formation process under photo-irradiation of the α-(2-naphthyl)-N-methylnitrone system. In the ONIOM extrapolation scheme,38–41 we can combine any two or three QM and MM methodologies into one calculation. In our present work, we have considered 2-layer QM:QM′ combination, which can be represented as CASSCF (4,4)/6-31G*:RHF/4-31G. The higher level quantum mechanical method is CASSCF and the lower level method is Hartree–Fock theory. This is expected to give a better interaction of the model part and the low-level system as both are treated at quantum mechanical level.46 The CASSCF-based higher level of theory has been treated with 4 active electrons in 4 active orbitals, considering the C–N–O moiety as the core region (model) of our photochemical interest which involves the C–N π bond and the pz orbital on oxygen in the nitrone–oxaziridine photo-conversion process. The total energy in the ONIOM methodology is expressed through an extrapolation scheme:
| EONIOM (QM, QM′) = EQM, model + EQM′, real − EQM′, model |
In addition to this hybrid method, a separate calculation has been also employed for the whole nitrone system at the CASSCF (4, 4) level of calculation. It has already been discussed that our prior experience on the photochemical study of the model N-alkyl retinyl nitrone system18 has clearly indicated that the involvement of orbitals having electronic cloud distributed over the portions of the molecule away from the CNO moiety in the active space does not lead to our desired oxaziridine product. The same is also expected to be true for the α-naphthyl N-methyl nitrone system, as experimental results indicate. This implies that inspite of employing huge computational cost by using a large active space in this present photochemical study it is less likely to obtain our desired nitrone–oxaziridine photo-conversion process. Therefore, we have attempted to make an accurate minimal choice of the active space leaving out the probable less important naphthyl part from it, and this method is completely biased towards the possible reaction path39 using previous intuition from our earlier work and the experimental findings of Kochany et al.23 In this CASSCF (4, 4) calculation the chosen HOMO is of π symmetry on the CNO moiety while the LUMO is of corresponding π* symmetry (Fig. S1†). It must be added that we had actually started our CASSCF calculations using a (14, 12) active space, but we were unable to find any oxaziridine type species on the photochemical path unlike the (4, 4) active space calculation. It is a fact that there is no single correct active space in a molecule;39 the choice of active space depends on the particular process being carried out. Our present theoretical investigation is based on a comparison of these CASSCF calculated parameters and the ONIOM-based CASSCF (4,4)/6-31G*:RHF/4-31G derived values. CAS perturbation theory (CASMP2) has introduced dynamic correlation effect of Moller–Plesset perturbation (MP2) level through single point calculations on top of the CASSCF optimized geometries. Transition states have been located using the normal TS technique based on the Berny-algorithm47 and QST3 (ref. 48) methodology. Intrinsic reaction coordinate (IRC) method49–51 has been used to follow the minimum energy path from the transition state.
In addition to these studies, GUGA-based configuration interaction singles and doubles (CISD) technique has also been used for some important calculations through the GAMESS52–56 suite of programs. Radiative transition57–59 calculations have been carried out between the two CI wavefunctions at the ground state equilibrium geometry, based on this GUGA CI code. Electrostatic potential-based atomic charges are calculated for the ground and excited state species using the Merz–Kollman60,61 scheme in Gaussian 09 program. For visualization of the output files, ChemCraft62 and Gaussview softwares have been employed throughout this computational work.
3. Results and discussion
3.1 Optimized ground state and excited state: structure, energy and atomic charges
Optimized ground and excited state geometries are compared at different level of calculations (Table 1, Fig. 1). The optimized ground state geometry of the Z-isomer is found to be more stable than the experimentally studied23 E-isomer of this compound (Table 2). Unlike the planar Z-isomer, the E-isomer is found to be non-planar where the naphthyl part is slightly tilted away from the plane of the double bond to avoid any steric interaction with the methyl group. In fact, all α-aryl N-alkyl nitrones are reported to have the Z-isomer as the stable form. The C–N double bond in the ground state becomes elongated in the first excited singlet state by roughly 0.1 Å (Fig. 1). A major difference has been observed in the planarity of the optimized excited state of the Z-isomer depending on the level of calculation employed; unlike the planar structure predicted by the ONIOM method, the CASSCF results (Fig. 1) have predicted a non-planar geometry of the excited singlet state with ∠C–C–N–O dihedral angle value of 57° for this isomer. Our rough estimation through a (4 × 4) PM3/CI calculation has also predicted a similar geometry (Fig. S2†) for the second CI root where the above-mentioned dihedral angle is roughly 43°. The ground and excited states of the unstable E-isomer are both characterized by non-planar geometries (Table 1, Fig. 1).| Molecular states | Level of calculationa | C–C | C–N | N–O | N–C | DC–C–C–N | DC–C–N–O |
|---|---|---|---|---|---|---|---|
| a Level of calculations are CASSCF (4,4)/6-31G* (level I), CASSCF (4,4)/6-31G*:RHF/4-31G (level II), CASSCF (14,12)/6-31G* (level III), RHF/6-311G** (level IV), B3LYP/6-311G**, (level V) and PM3/CI (level VI). | |||||||
| Ground state of Z-isomer | I | 1.469 | 1.311 | 1.276 | 1.459 | 0.0 | 0.0 |
| II | 1.456 | 1.291 | 1.319 | 1.455 | 0.0 | 0.0 | |
| III | 1.465 | 1.303 | 1.265 | 1.462 | 0.0 | 0.0 | |
| IV | 1.465 | 1.276 | 1.265 | 1.462 | 0.0 | 0.0 | |
| V | 1.447 | 1.318 | 1.271 | 1.482 | 0.0 | 0.0 | |
| VI | 1.455 | 1.334 | 1.243 | 1.507 | −24.2 | 0.1 | |
| Ground state of E-isomer | I | 1.483 | 1.307 | 1.254 | 1.466 | −60.0 | 176.6 |
| II | 1.473 | 1.296 | 1.274 | 1.495 | −50.5 | 176.8 | |
| III | 1.481 | 1.307 | 1.254 | 1.466 | −57.5 | 176.6 | |
| IV | 1.481 | 1.274 | 1.265 | 1.465 | −55.7 | 177.4 | |
| V | 1.458 | 1.318 | 1.270 | 1.483 | −34.7 | 175.5 | |
| VI | 1.460 | 1.330 | 1.247 | 1.500 | −49.1 | −179.7 | |
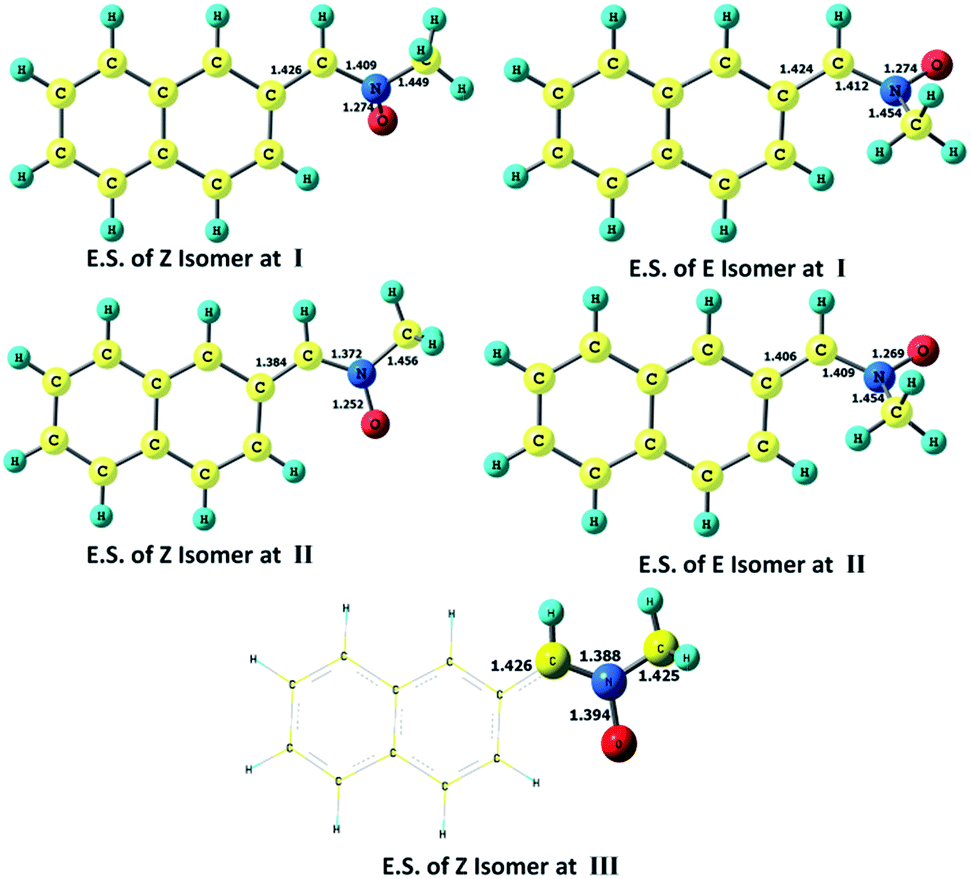 | ||
| Fig. 1 Optimized excited state geometries of E and Z isomers at CASSCF (4,4)/6-31G* (I), CASSCF (14,12)/6-31G* (II) and ONIOM (QM:QM′) (III) level of calculations. | ||
| Molecular state | Level of calculationa | Z-isomer | E-isomer |
|---|---|---|---|
| ΔE in kcal mol−1 | ΔE in kcal mol−1 | ||
| a Level of calculations are CASSCF (4,4)/6-31G* (level I), CASMP2 (4,4)/6-31G* (level II), CASSCF (4,4)/6-31G*:RHF/4-31G (level III) and CASSCF (14,12)/6-31G* (level IV).b Experimental value is 327 nm (ref. 23). | |||
| G. S. | I | 0 | 0 |
| II | 0 | 0 | |
| III | 0 | 0 | |
| IV | 0 | 0 | |
| E. S. | I | 110.68 | 88.62 |
| II | 102.05 | 84.52 | |
| III | 86.62 | — | |
| IV | 84.38 | 82.70 | |
| F. C. | I | 118.31 (241) | 91.30 (313)b |
| II | 110.89 (257) | 84.83 (337)b | |
| III | 98.02 (291) | 102.34 (279)b | |
| IV | 95.83 (298) | 103.49 (276)b | |
The calculated vertical excitation energies (Table 2) of the Z-isomer from the CASSCF method with (4, 4) and (14, 12) active spaces are 118 and 96 kcal mol−1, respectively. The CASMP2 calculations have predicted a value of 110 kcal mol−1 while the 2-layer ONIOM method has given a value (98 kcal mol−1) close to the calculated values at CASSCF (14, 12) level. These latter values have predicted an absorption peak position close to 295 nm. On the other hand, the VEE values predicted at the CASSCF (91 kcal mol−1, 315 nm) and CASMP2 (85 kcal mol−1, 335 nm) level using (4, 4) active space for the E-isomer are substantially lower than the CASSCF (14, 12) and the ONIOM predicted VEE values (102–103 kcal mol−1) for this isomer. The experimentally reported peak position23 of this isomer is close to 325 nm. After initial photo-excitation, the vertically excited state relaxes to a comparatively stable geometry which is situated at 8–12 kcal mol−1 below the excited state FC geometry for the Z-isomer. However, in case of the E-isomer this stabilization is smaller (1–2 kcal mol−1). The CASSCF calculated ESP-based atomic charges (Table S1†) are quite similar to the values observed in our earlier studied long-chain conjugated nitrone systems. A clear indication of initial electronic transfer from the oxygen atom to the nitrogen can be seen as the relaxation towards the optimized excited state is approached after the initial photo-excitation; this electronic transfer triggers the subsequent photochemical processes.
3.2 Non-radiative decay channel
In the next stage of our work we have searched the probable conical intersection and transition state geometries on the excited state surface. Some rough estimation through the semi-empirical (PM3) configuration interaction method (mentioned in ESI†) has been employed to have some idea of the starting guess structures for the conical intersection geometries. These guess geometries are then utilized for the conical intersection optimization run through the CASSCF and ONIOM level of calculations.The initial guess geometry corresponding to the Z-isomer has lead to two different types of conical intersections at the higher level of ab initio calculations. Rotation around the C–C bond and C–N bond causes these two types of intersection points. The one with simultaneous change of the C–C–C–N and C–C–N–O dihedral angles (Table 3) gives rise to the conical intersection point CI1, which seems to originate from a hula twist (HT)-type motion, normally responsible for the kinked conical intersection in conjugated polyenes. On the other hand, the terminally-twisted CI2 intersection geometry (Fig. 2) resembles the lowest-energy conical intersections earlier reported by us for the long-chain conjugated nitrone systems. This latter geometry involves an out-of-plane C–N–O kink or oxygen-bridge structure due to a rotation about the C–N bond resulting in a change in the C–C–N–O dihedral angle. Both of them are having almost similar atomic charges and these are close to the values reported for the lowest energy CI geometry for the model retinyl nitrone systems. In other words, similar to the retinyl nitrone systems, the ESP-derived charges for this α-naphthyl N-methylnitrone indicate biradicaloid conical intersection geometries with an odd electron on the α-C and a reduced electronic cloud on oxygen with a lone pair on nitrogen. Though the geometries obtained from the CI optimization run from the two different methods are apparently similar, the predicted C–C and C–N bond lengths differ by 0.05 Å. The hybrid method has predicted these structures at 20–26 kcal mol−1 below (Table 4) the optimized excited state while the CASSCF and CASMP2 methods have predicted these values between 33 and 38 kcal mol−1. In both the cases (CI1 and CI2), the gradient difference vectors indicate (Fig. 2) the possibility of a C–N–O triangle formation. Following these vectors by shortening the C–O distance we have obtained optimized ground state geometries (Fig. 3) in both the cases (Ox1 and Ox2) with a CNO kink having bond lengths and bond angles closely resembling the reported parameters of oxaziridine structure.63 The CASSCF predicted values of geometrical parameters of Ox1 and Ox2 (RC–O = 1.38 Å, RN–O = 1.44 Å, RC–N = 1.40 Å, ∠OCN = 62°, ∠ONC = 58°) are close to the hybrid ONIOM-predicted values of these two geometries, with some exceptions in the C–O and N–O bond distances. The experimentally reported oxaziridine geometry has a C–N–O triangle with C–O and N–O bond lengths of 1.40 Å and 1.50 Å, respectively, and the C–N bond length is approximately 1.44 Å; the bond angles ∠OCN = 63.7° and ∠ONC = 56.8° are also close to our predicted results. Energy-wise, the CASSCF and CASMP2 values suggest that these oxaziridine geometries are situated between the optimized ground and excited states; on the other hand, the ONIOM study has located their positions below the starting ground state geometry. Molecular orbital analysis of both these structures clearly gives the evidence of C–O bond formations. In fact, from the figure of HOMO, a three-centered C–O–N-type of bond formation (Fig. 3) is clearly visible in Ox1 and Ox2.
| Molecular geometry | Level of calculation | RC–C | RC–N | RN–O | RC–O | RN–C | DC–C–C–N | DC–C–N–O |
|---|---|---|---|---|---|---|---|---|
| CI1 | I | 1.453 | 1.392 | 1.360 | 2.125 | 1.445 | 176.0 | −80.5 |
| II | 1.392 | 1.474 | 1.366 | 2.355 | 1.461 | −174.5 | −116.5 | |
| CI2 | I | 1.464 | 1.389 | 1.387 | 2.128 | 1.444 | 17.2 | −71.0 |
| II | 1.413 | 1.446 | 1.385 | 2.227 | 1.446 | −1.0 | −79.2 | |
| CI3 | I | 1.376 | 1.466 | 1.257 | 2.340 | 1.450 | −173.1 | −168.8 |
| II | — | — | — | — | — | — | — | |
| CI4 | I | 1.453 | 1.409 | 1.368 | 2.173 | 1.446 | 8.0 | 82.0 |
| II | 1.428 | 1.428 | 1.406 | 2.206 | 1.448 | 3.0 | 78.1 | |
| Ox1 | I | 1.493 | 1.406 | 1.443 | 1.378 | 1.449 | −125.1 | −108.9 |
| II | 1.486 | 1.405 | 1.425 | 1.529 | 1.450 | −135.1 | −109.6 | |
| Ox2 | I | 1.490 | 1.404 | 1.442 | 1.381 | 1.450 | 36.8 | −108.8 |
| II | 1.484 | 1.403 | 1.528 | 1.427 | 1.450 | 36.7 | −109.5 | |
| Ox3 | I | 1.490 | 1.404 | 1.404 | 1.384 | 1.450 | −36.7 | 108.8 |
| II | 1.483 | 1.403 | 1.427 | 1.528 | 1.450 | 36.7 | 109.5 |
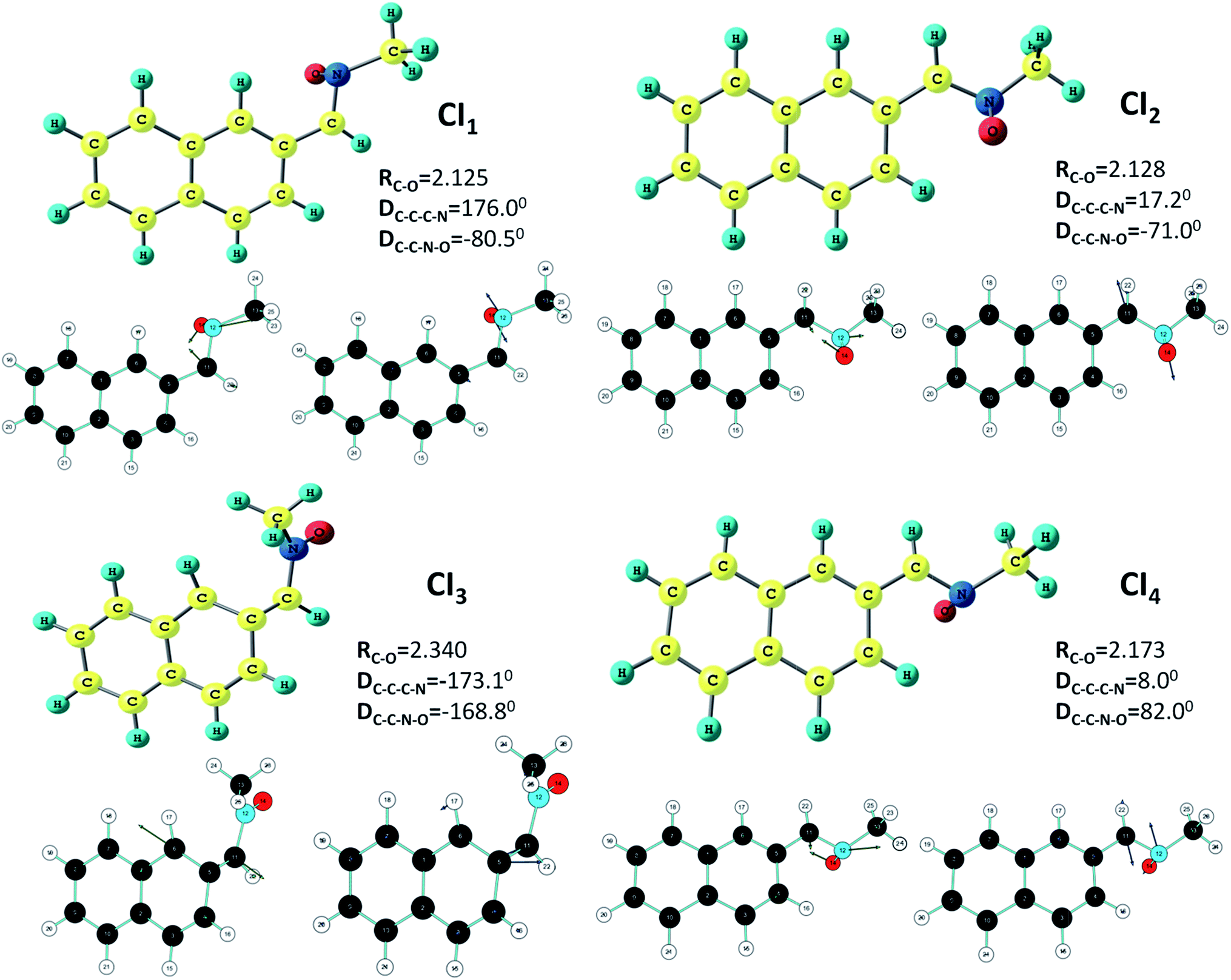 | ||
| Fig. 2 Optimized conical intersection geometries (CI1, CI2, CI3 and CI4) at the CASSCF/6-31G* level. The corresponding gradient difference and derivative coupling vectors are also shown. | ||
| Molecular geometry | CASSCF | CASMP2 | ONIOM (CAS:RHF) |
|---|---|---|---|
| ΔE in kcal mol−1 | ΔE in kcal mol−1 | ΔE in kcal mol−1 | |
| E.S. (Z isomer) | 0 | 0 | 0 |
| E.S. (E isomer) | 0.20 | 0.80 | — |
| CI1 | −35.49 | −36.29 | −20.34 |
| CI2 | −33.50 | −37.68 | −26.15 |
| CI3 | 12.31 | 10.27 | — |
| CI4 | −41.52 | −36.87 | −27.23 |
| Ox1 | −81.49 | −93.57 | −91.27 |
| Ox2 | −82.68 | −94.22 | −92.20 |
| Ox3 | −96.96 | −93.61 | −92.20 |
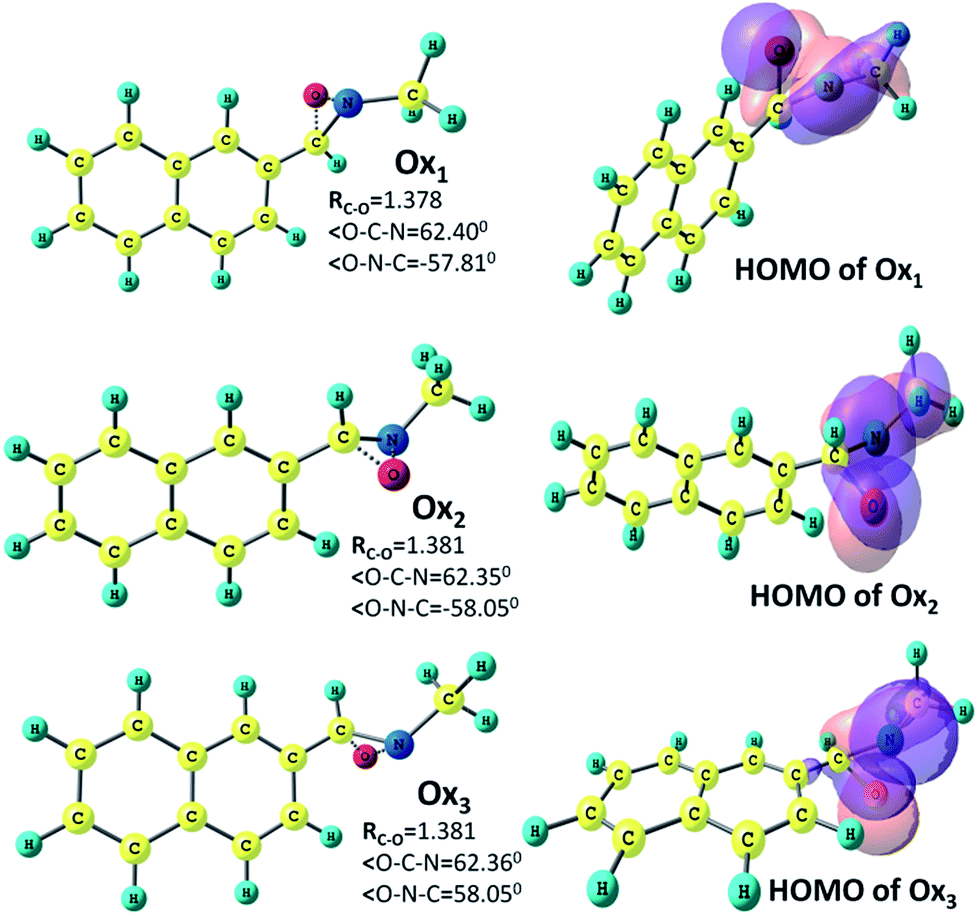 | ||
| Fig. 3 Optimized geometries of the oxaziridines (Ox1, Ox2 and Ox3) at the CASSCF/6-31G* level. The HOMOs of the corresponding geometries are also shown. | ||
Following the guess structure provided by the semi-empirical configuration interaction result for the E-isomer, two conical intersection geometries, namely CI3 and CI4, are obtained at the CASSCF and ONIOM level of calculations. The first one (CI3) has a twist in the opposite direction of CI1, however, situated at very high-energy level; on the other hand, the energy of the low-lying CI4 intersection point with a terminal C–N–O kink on the backside is found to be close to the CI2 geometry at the CASMP2 and ONIOM level, though CASSCF energies have predicted this geometry (CI4) as the lowest-energy CI point. This conical intersection point is situated at 60–69 kcal mol−1 (Table 4) above the ground state optimized geometry of the Z-isomer. The turn of the C–C–N–O dihedral angle in this species is exactly in the reverse direction in comparison to that of the CI2 geometry. The CASSCF and ONIOM-based geometrical parameters are in close agreement with each other, while the latter method has predicted this geometry at a higher energy value (51 kcal mol−1) than the CASSCF and CASMP2 reported values (47 kcal mol−1) with respect to the ground state geometry of E-isomer. The gradient difference vectors of this conical intersection point is clearly showing a chance of C–N–O triangle formation through a C–O bond. Following these vectors we have obtained an oxaziridine geometry (Ox3) which almost matches the reported geometrical parameters of this 3-membered heterocyclic species. Unlike the other two oxaziridine structures, the Ox3 species is situated below (8.5–9.9 kcal mol−1) the ground state geometry of the E-isomer at the CASSCF and CASMP2 level of studies, while the hybrid method has predicted its location at 13.7 kcal mol−1 lower than this ground state geometry. Similar to the other two oxaziridine structures, a clear overlap of orbitals on carbon and oxygen can be noticed from the picture of HOMO of Ox3 geometry along with the formation of a 3-centred C–O–N bond. Though the oxaziridine geometry Ox3 and the conical intersection CI4 are mentioned to be arising from the E-isomer based on the rough estimation of semi-empirical guess structures, it must be added that these two geometries may originate from the Z-isomer, as well. In fact, the back-side turn of the C–C–N–O dihedral angle (57.3°) in the optimized excited state of the Z-isomer is also quite consistent with the formation of the CI4 geometry where the angle increases to 82°.
We have tried to analyze the reason behind the difference in stabilities of the three important conical intersection geometries (CI1, CI2, and CI4). The Hula-twist motion involved in the CI1 geometry is known to be a volume demanding process and therefore its energy is expected to be higher. On the other hand, the CI2 and CI4 geometries have terminal-twist (OBF-type) with C–C–N–O turn in the front and back sides, respectively, which requires less volume change. However, the energy gaps between the CI1 geometry and the other two low-lying CI geometries are not very high; this is due to the fact that unlike our previously studied conjugated-chain N-alkyl nitrone systems, there exists an additional strain in this presently investigated nitrone system due to the naphthyl ring and the methyl group/oxygen atom, when they come to close vicinity. As a consequence, the out-plane Hula twist motion though expected to be at significantly high energy level, seems to be sterically more favored, and therefore energy-wise not too different from the terminally twisted structures.
3.3 Optimized transition states on the excited state and ground state surfaces
Two transition states (TSex1 and TSex2) have been optimized in the excited state for the Z-isomer at the CASSCF level of calculations (Fig. S3†). The planar TSex2 has a negative frequency of 126 cm−1 and the vectors corresponding to this frequency indicates a turn of the oxygen atom towards the front side. It seems that following this frequency we can reach the conical intersection geometry CI2 which subsequently leads to Ox2. The other optimized TS geometry (TSex1) has a slightly higher negative frequency (175 cm−1) and not directly connected to this photochemical path. A transition state with almost similar geometrical parameters (TSex3) with an opposite C–N–O twist has been obtained from the E-isomer which possesses a negative frequency of 139 cm−1. All these excited state TS geometries are situated marginally (1–2 kcal mol−1) above the optimized excited state geometries.A similar investigation of transition states on the ground state surface of the Z-isomer has led to two TS geometries (Fig. 4) at CASSCF level (TSgs1 and TSgs2) while two different transition states have been obtained at the ONIOM level (TSgs3 and TSgs4). The only negative frequency of TSgs2 which indicates stretching and contracting along the C5–O bond is 1305 cm−1. This is consistent with the IR stretching frequency of the C–O bond and there is enough reason to conclude that this TS connects the oxaziridine (Ox2) and the Z-isomer of the nitrone, and indicates that the C5–O bond will open up to give back the parent nitrone passing through this transition state. The ONIOM-predicted TSgs3 and TSgs4 structures are characterized by negative frequencies of 332 cm−1 and 323 cm−1, respectively. In both the cases the vectors are clearly showing chances of C–O bond formation. It must be added here that the gradient difference vectors of CI1 and CI2 are parallel to the vectors corresponding to the negative frequencies of TSgs3 and TSgs4, respectively, and both seems to be leading towards their respective oxaziridine structures. The TSgs3 geometry is situated at 40 kcal mol−1 below the CI1 intersection point and 30 kcal mol−1 above the Ox1 geometry, while the TSgs4 geometry lies half-way between the CI2 and the Ox2 geometries. On the whole, both these transition states are situated on the nitrone–oxaziridine photochemical conversion pathway.
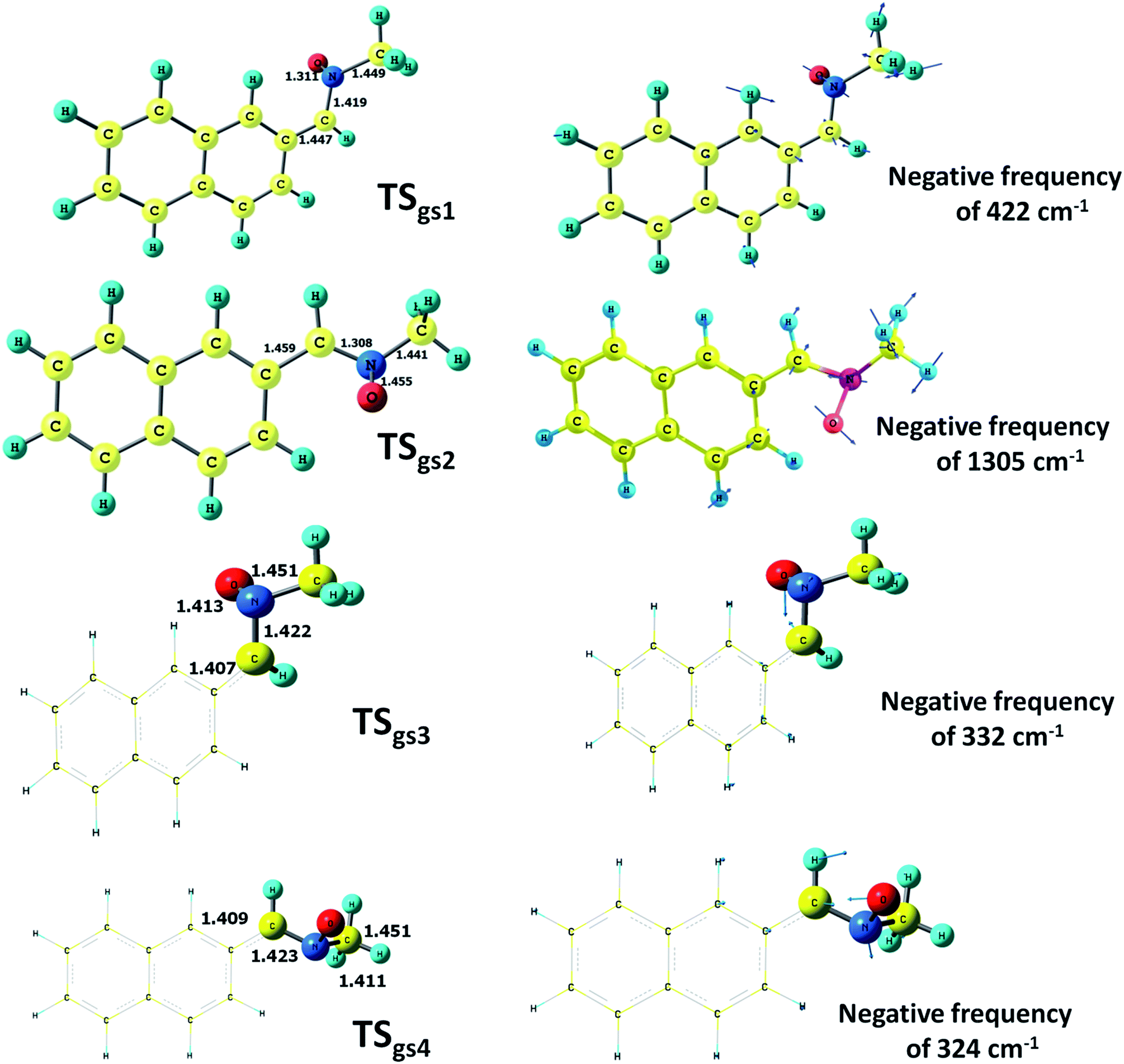 | ||
| Fig. 4 Optimized transition states geometries (TSgs1-4) on ground state surface with displacement vectors of their respective negative frequencies. | ||
The fifth transition state detected on the ground state surface (TSgs5) has been identified as the link between the E–Z isomers (Fig. 5). The QST3 methodology in Gaussian has been employed for finding out this transition state where the E-isomer has been specified as the reactant molecule while the Z-isomer is taken as the product. This transition state, which is responsible for the E–Z thermal isomerization, is characterized by a negative frequency of 350 cm−1. The unstable E isomer which is situated at 22 kcal mol−1 above the stable Z-isomer, reaches this transition state with activation energy of 23.7 kcal mol−1 (Table S2†). Thereafter following the negative frequency, the Z-isomer is formed. This optimized ground state of the latter isomer is situated at 46 kcal mol−1 below the TSgs5 geometry. An IRC run has been given on this transition state both in the forward and reverse directions; the corresponding IRC plot is shown in Fig. S4.† It should be mentioned here that in recent times E–Z isomerization studies64,65 on different types of nitrones have been extensively reported at the DFT level of calculations.
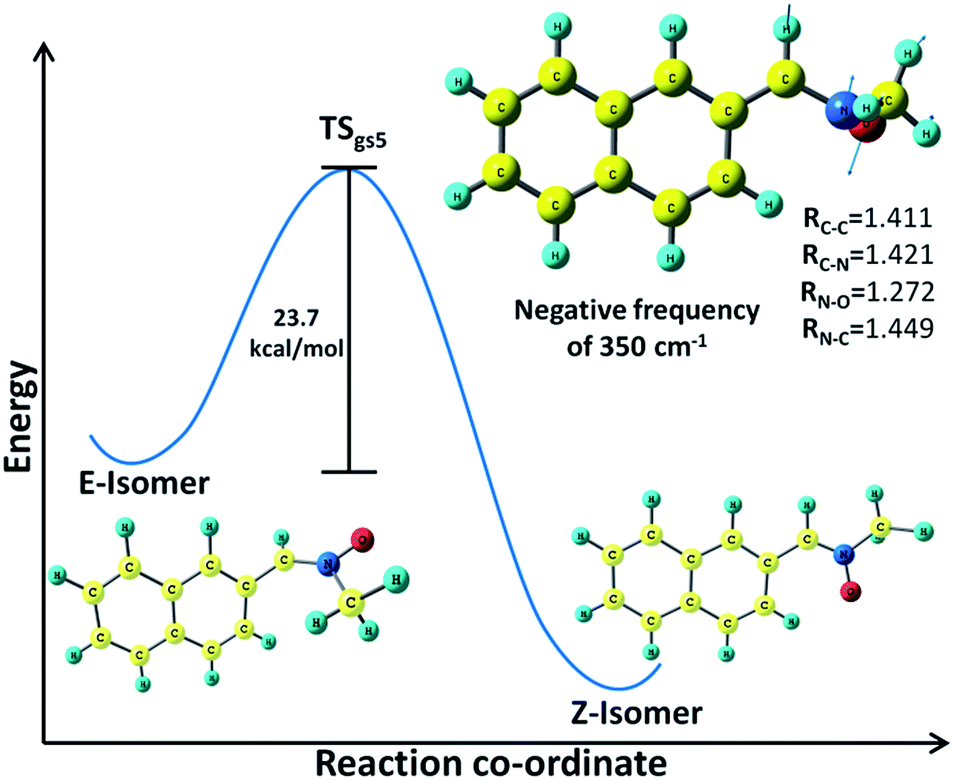 | ||
| Fig. 5 Schematic representation of E–Z isomerization of α-(2-naphthyl)-N-methylnitrone through TSgs5. | ||
3.4 Radiative transition studies
Unlike our previously studied retinyl nitrones, this presently investigated α-naphthyl N-methyl nitrone systems are reported to give fluorescence emission.23 We have attempted a radiative transition analysis on the low-lying S0–S1, S0–S2, S2–S1 transitions and their corresponding properties through calculations based on GUGA CISD code in GAMESS. These radiative transition related studies are carried out at the ground state equilibrium geometry from where the vertical transition to the excited state (Franck Condon geometry) originates. This study reveals that the transition to the first excited singlet state is very weak (TM value: 0.30 Debye for Z-isomer and 0.37 Debye for E-isomer) for both the isomers with low oscillator strength values, and the corresponding Einstein coefficient (A) values are in the order of 107 (Table 5). The radiative lifetime values of the S1 states are expected to be in the order of nanoseconds (roughly 35–60 ns) for these isomers. On the other hand, the vertical transition strengths to the second excited singlet states (S2) at the ground state geometries are significantly strong (TM value: 5.61 Debye for Z-isomer and 3.862 Debye for E-isomer) with high oscillator strength values and Einstein coefficients in the order of 109 which corresponds to radiative lifetime values of pico second order (roughly 140–275 ps). The S2–S1 transition moments are also moderately strong with TM values of 2.0 Debye with oscillator strengths of 0.01. These results are quite different from our previously studied long-chain conjugated N-alkyl substituted nitrones where the S0–S1 transition was found to be the strongest (TM value of 4.86 Debye and oscillator strength value of 0.71) while their S0–S2 transition was very weakly allowed with low transition moment value. Radiative transition result of this long-chain conjugated N-alkyl nitrone was more towards the conjugated iminium ion systems. Properties of this nitrone were reported to be somewhat in between the conjugated non-polar polyenes and the structurally similar long-chain iminium ion systems. However, results of our presently studied nitrone system indicates its similarity with the conjugated methyl-substituted hexatriene system (Fig. 6) where the S0–S1 transition has slightly more transition moment value (TM value 0.97 D) and the S0–S2, S2–S1 transitions are strongly allowed. In conjugated non-polar polyene systems, the initial transition to S2 (ionic) state is followed by a quick downward transition to the S1 (biradical) state before reaching the ground state (S0) which eventually gets involved in a kinked conical intersection with this S0 state through a Hula-twist motion. A similar low-lying kinked CI (CI1) is also observed in our presently analyzed α-naphthyl N-methyl nitrone system; however, the less volume demanding terminally-twisted CI's are also obtained at low-energy level which is a characteristic of the photo-excited nitrone system leading towards the oxaziridine geometry.| Systems | Transition moment in Debye | Oscillator strength | Einstein's coefficient in s−1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| S0–S1 | S0–S2 | S2–S1 | S0–S1 | S0–S2 | S2–S1 | S0–S1 | S0–S2 | S2–S1 | |
| a The values in parentheses are the powers to the base 10. | |||||||||
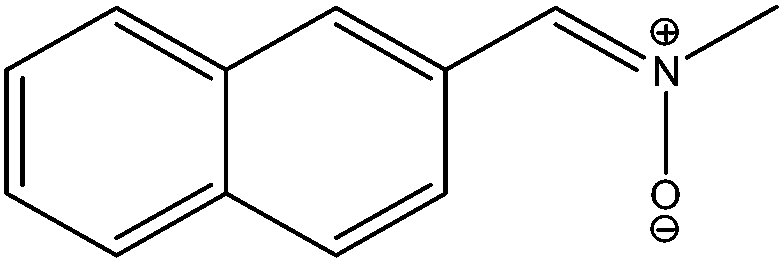 |
0.304 | 5.616 | 2.099 | 0.003 | 1.322 | 0.010 | 1.7280 (+7) | 7.0138 (+9) | 1.8008 (+5) |
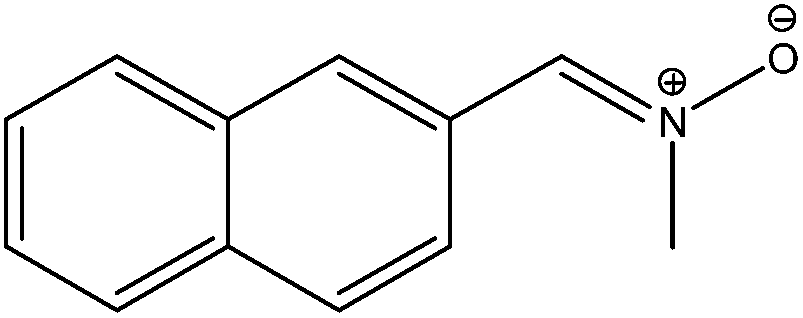 |
0.371 | 3.862 | 1.890 | 0.005 | 0.644 | 0.010 | 2.7374 (+7) | 3.6303 (+9) | 2.5472 (+5) |
 |
0.961 | 7.764 | 4.303 | 0.028 | 1.861 | 0.010 | 7.7773 (+7) | 5.3540 (+9) | 9.1551 (+3) |
 |
4.861 | 0.112 | 0.069 | 0.706 | 0.000 | 0.000 | 2.1472 (+9) | 1.0559 (+6) | 1.2264 (+0) |
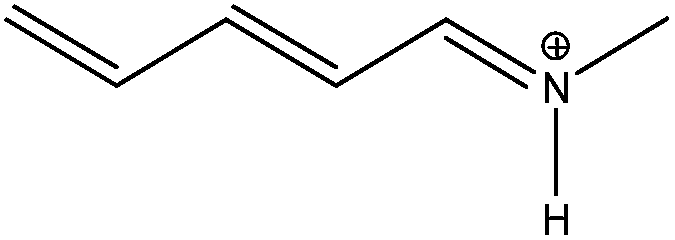 |
7.543 | 1.891 | 1.935 | 1.695 | 0.109 | 0.019 | 4.5380 (+9) | 3.1182 (+8) | 1.5590 (+6) |
 | ||
| Fig. 6 Schematic representation of the low-lying singlet–singlet transitions and involvement of some important low-lying conical intersections (S0/S1) in α-(2-naphthyl)-N-methylnitrone system. | ||
A comparative analysis of the four above-mentioned conjugated systems has revealed that the vertically excited S1 state of 2,4-pentadien-1-iminium ion is completely dominated by the configuration arising due to the HOMO → LUMO excitation (Table S3†) while it partly dominates this state in the conjugated N-methyl nitrone system. In contrast, this configuration dominates the S2 state of the 1,3,5-hexatriene system and our presently studied α-naphthyl N-methyl nitrone system at the FC geometry. The S1 states of these latter systems are mostly contributed by configurations arising due to the HOMO → LUMO + 1 and HOMO − 1 → LUMO excitations at the above-mentioned geometries. The conjugated non-polar polyene has also a major contribution from the HOMO2 → LUMO2 configuration for this first vertically excited singlet state (at FC geometry). Interestingly, unlike in the iminium ion, this doubly excited configuration becomes a key player in the S1 state of the nitrones and the polyenes as the optimized geometry of this state is approached. This optimized excited state in the α-naphthyl N-methyl nitrone is almost equally contributed by configurations due to HOMO2 → LUMO2, HOMO → LUMO + 1 and HOMO − 1 → LUMO excitations (Table S3†).
Experimental studies have revealed23 that the photoproduct oxaziridines are also fluorescent in nature which results in increase and fluctuation of fluorescence intensity of the parent nitrone system during their irradiation. GUGA CISD-based radiative transition studies have been carried out on the ground state oxaziridine systems and the results (Table S4†) are found to be quite similar to those of the parent nitrone systems. The S0–S1 transitions are weaker than the S0–S2 and S2–S1 transitions; however, the transition moment and oscillator strength values of the latter-mentioned transitions are significantly lower in comparison to those values of nitrones. On the other hand, these parameters of the lowest transition (S0–S1) are found to decrease slightly in this heterocyclic species.
4. Conclusion
This computational study has reported a comprehensive analysis of the photochemical and photophysical properties of the α-(2-naphthyl)-N-methylnitrone system. The photochemical nitrone–oxaziridine conversion is found to involve biradicaloid conical intersection geometries through low-lying Hula-twist and terminal one-bond flip motions. The optimized oxaziridine geometries closely resemble the previously reported geometry of this three-membered heterocyclic system and presence of a 3-centred molecular orbital is clearly observed in this species. The transition state responsible for the thermal E–Z isomerization of this nitrone has also been identified in this present work. In addition to these, the radiative transition studies of this nitrone have revealed some interesting findings, too. Unlike our previously reported conjugated long-chain N-methyl nitrones, the vertical transition to the first excited singlet state is weakly allowed in this system. This excited state may be also populated by downward transition from the second excited singlet state and eventually gets involved in the S0/S1 conical intersections to produce oxaziridine and other photoproducts. This behavior is almost similar to that of the non-polar conjugated polyene systems. On the other hand, the transition properties of the long-chain conjugated nitrones were found to be somewhat close to that of the structurally similar iminium ion systems. It establishes the fact that properties of nitrones are in between the polyenes and iminium ion systems. The predicted absorption peak position of the E isomer of this nitrone almost matches its experimentally reported value, and its excited singlet state which was reported to give fluorescence is found to have a radiative lifetime of nanosecond order from the Einstein's coefficient values. Overall, these computed results at various level of quantum mechanical calculations are expected to contribute significantly to the properties of the α-(2-naphthyl)-N-methylnitrone. However, it must be concluded that the importance of this work is not only restricted to this particular type of nitrone system rather it can have far reaching consequences in terms of understanding the inner details of the photochemistry and isomerization processes of several other structurally similar nitrone systems, too.Acknowledgements
We gratefully acknowledge the financial support received from the Council of Scientific and Industrial Research (CSIR), Government of India, under the Scheme no. 01(2681)/12/EMR-II, for the present work.Notes and references
- M. A. Saliva and J. M. Goodman, Tetrahedron, 2002, 58, 3667 CrossRef.
- K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed.
- A. Padwa and G. S. K. Wong, J. Org. Chem., 1986, 51, 3125 CrossRef CAS.
- E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1969, 91, 4481 CrossRef CAS.
- R. A. Floyd, K. Hensley, M. J. Forster, J. A. Kelleher-Anderson and P. L. Wood, Ann. N. Y. Acad. Sci., 2002, 959, 321 CrossRef CAS PubMed.
- F. A. Villamena, A. Das and K. M. Nash, Future Med. Chem., 2012, 4, 1171 CrossRef CAS PubMed.
- R. A. Floyd, R. D. Kopke, C.-H. Choi, S. B. Foster, S. Doblas and R. A. Towner, Free Radical Biol. Med., 2008, 15, 1361 CrossRef PubMed.
- R. A. Floyd, K. Hensley, M. J. Forster, J. A. Kelleher-Anderson and P. L. Wood, Mech. Ageing Dev., 2002, 123, 1021 CrossRef CAS.
- R. A. Floyd, H. C. C. F. Neto, G. A. Zimmerman, K. Hensley and R. A. Towner, Free Radical Biol. Med., 2013, 62, 145 CrossRef CAS PubMed.
- A. Samadi, E. Soriano, J. Revuelta, C. Valderas, M. Chioua, I. Garrido, B. Bartolomé, I. Tomassolli, L. Ismaili, L. González-Lafuente, M. Villarroya, A. G. García, M. J. Oset-Gasque and J. Marco-Contelles, Bioorg. Med. Chem., 2011, 19, 951 CrossRef CAS PubMed.
- M. Chioua, D. Sucunza, E. Soriano, D. Hadjipavlou-Litina, A. Alcázar, I. Ayuso, M. J. Oset-Gasque, M. P. González, L. Monjas, M. I. Rodríguez-Franco, J. Marco-Contelles and A. Samad, J. Med. Chem., 2012, 55, 153 CrossRef CAS PubMed.
- R. A. Floyd, R. A. Towner, D. Wu, A. Abbott, R. Cranford, D. Branch, W.-X. Guo, S. B. Foster, I. Jones, R. Alam, D. Moore, T. Allen and M. Huycke, Free Radical Res., 2010, 44, 108 CrossRef CAS PubMed.
- B. Guo, D. Xu, H. Duan, J. Du, Z. Zhang, S. M. Lee and Y. Wang, Biol. Pharm. Bull., 2014, 37, 274 CAS.
- R. A. Floyd, H. K. Chandru, T. He and R. Towner, Anti-Cancer Agents Med. Chem., 2011, 11, 373 CrossRef CAS.
- P. Astolfi, P. Carloni, M. G. Marini, G. Mobbili, M. Pisani and P. Stipa, RSC Adv., 2013, 3, 22023 RSC.
- X. Song, Y. Qian, R. Ben, X. Lu, H.-L. Zhu, H. Chao and J. Zhao, J. Med. Chem., 2013, 56, 6531 CrossRef CAS PubMed.
- V. Balogh-Nair and K. Nakanishi, Pharm. Res., 1984, 1, 93 CrossRef CAS.
- P. Saini and A. Chattopadhyay, RSC Adv., 2014, 4, 20466 RSC.
- J. S. Splitter and M. Calvin, J. Am. Chem. Soc., 1965, 30, 3427 CAS.
- J. S. Splitter, T.-M. Su, H. Ono and M. Calvin, J. Am. Chem. Soc., 1971, 93, 4075 CrossRef CAS.
- K. Shinzawa and I. Tanaka, J. Phys. Chem., 1964, 68, 1205 CrossRef CAS.
- K. Koyano and I. Tanaka, J. Phys. Chem., 1965, 69, 2545 CrossRef CAS.
- E. Lipczynska-Kochany and J. Kochany, J. Photochem. Photobiol., A, 1988, 45, 65 CrossRef CAS.
- W. Domcke and D. R. Yarkony, Annu. Rev. Phys. Chem., 2012, 63, 325 CrossRef CAS PubMed.
- J. H. Lehman and M. I. Lester, Annu. Rev. Phys. Chem., 2014, 65, 537 CrossRef CAS PubMed.
- S. Gozem, E. Mirzakulova, I. Schapiro, F. Melaccio, K. D. Glusac and M. Olivucci, Angew. Chem., Int. Ed., 2014, 53, 9870 CrossRef CAS PubMed.
- H. Timmers, Z. Li, N. Shivaram, R. Santra, O. Vendrell and A. Sandhu, Phys. Rev. Lett., 2014, 113, 113003 CrossRef.
- T. Mori and T. J. Martínez, J. Chem. Theory Comput., 2013, 9, 1155 CrossRef CAS.
- D. R. Yarkony, Chem. Rev., 2012, 112, 481 CrossRef CAS PubMed.
- A. Migani and M. Olivucci, Conical Intersections: Electronic Structure, Dynamics & Spectroscopy Advanced series in Physical Chemistry, ed. W. Domcke, D. R. Yarkony and H. Koppel, World Scientific Publishing Co. (P). Ltd, Singapore, 2004, vol. 15 Search PubMed.
- I. J. Palmer, I. N. Ragazos, F. Bernardi, M. Olivucci and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 673 CrossRef CAS.
- J. Dreyer and M. Klessinger, Chem.–Eur. J., 1996, 2, 335 CrossRef CAS.
- N. Yamamoto, T. Vreven, M. A. Robb, M. J. Frisch and H. B. Schlegel, Chem. Phys. Lett., 1996, 250, 373 CrossRef CAS.
- M. J. Frisch, I. N. Ragazos, M. A. Robb and H. B. Schlegel, Chem. Phys. Lett., 1992, 189, 524 CrossRef CAS.
- S. Dapprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 462, 1 CrossRef.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery Jr, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815 CrossRef CAS.
- T. Vreven and K. Morokuma, Ann. Rep. Comput. Chem., 2006, 2, 35 CrossRef CAS.
- T. Vreven, K. Morokuma, O. Farkas, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 2003, 24, 760 CrossRef CAS PubMed.
- M. J. Bearpark, F. Ogliaro, T. Vreven, M. Boggio-Pasqu, M. J. Frisch, S. M. Larkin, M. Morrison and M. A. Robb, J. Photochem. Photobiol., A, 2007, 190, 207 CrossRef CAS PubMed.
- M. J. Bearpark, S. M. Larkin and T. Vreven, J. Phys. Chem. A, 2008, 112, 7286 CrossRef CAS PubMed.
- A. Bhattacharya and E. R. Bernstein, J. Phys. Chem. A, 2011, 115, 4135 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- P. Slavíček and T. J. Martínez, J. Chem. Phys., 2010, 132, 234102 CrossRef PubMed.
- J. J. W. McDouall, K. Peasley and M. A. Robb, Chem. Phys. Lett., 1988, 148, 183 CrossRef CAS.
- J. F. Arenas, J. I. Marcos, J. C. Otero and A. Sanchez-Galvez, J. Chem. Phys., 1999, 111, 551 CrossRef CAS PubMed.
- M. Lundberg, J. Comput. Chem., 2012, 33, 406 CrossRef CAS PubMed.
- X. Li and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 835 CrossRef CAS.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, Theory and Applications of Computational Chemistry: The First 40 Years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. Scuseria, Elsevier, Amsterdam, 2005 Search PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. J. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- B. Brooks and H. F. Schaefer, J. Chem. Phys., 1979, 70, 5092 CrossRef CAS PubMed.
- B. Brooks, W. Laidig, P. Saxe, N. Handy and H. F. Schaefer, Phys. Scr., 1980, 21, 312 CrossRef CAS.
- A. Chattopadhyay, J. Phys. B: At., Mol. Opt. Phys., 2012, 45, 165101 CrossRef.
- A. Chattopadhyay, J. Chem. Sci., 2012, 124, 985 CrossRef CAS.
- F. Weinhold, J. Chem. Phys., 1970, 54, 1874 CrossRef PubMed.
- C. W. Bauschlicher and S. R. Langhoff, Theor. Chim. Acta, 1991, 79, 93 CrossRef CAS.
- S. Koseki and M. S. Gordon, J. Mol. Spectrosc., 1987, 123, 392 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129 CrossRef CAS.
- B. H. Besler, K. M. Merz Jr and P. A. Kollman, J. Comput. Chem., 1990, 11, 431 CrossRef CAS.
- Website of Chemcraft software, http://www.chemcraftprog.com.
- S. S. Murphree, Modern Heterocyclic Chemistry, ed. J. Alvarez- Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH Verlag & Co. KGaA, Weinhelm, Germany, 2011 Search PubMed.
- L. Meng, S. C. Wang, J. C. Fettinger, M. J. Kurth and D. J. Tantillo, Eur. J. Org. Chem., 2009, 1578 CrossRef CAS PubMed.
- D. Roca-López, T. Tejero and P. Merino, J. Org. Chem., 2014, 79, 8358 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Orbitals involved in the (4, 4) active space in CASSCF/6-31G* calculations; Fig. S1; semi-empirical CI (PM3/CI) level of studies; Fig. S2; ESP-derived atomic charges using Merz–Kollman scheme; Table S1; transition states; Fig. S3, S4 and Table S2; dominant configurations of S1 and S2 states Tables S2 and S3; Cartesian coordinates. See DOI: 10.1039/c4ra16375c |
| This journal is © The Royal Society of Chemistry 2015 |