
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibacterial wound dressing: plasma treatment effect on chitosan impregnation and in situ synthesis of silver chloride on cellulose surface
Vladimíra
Vosmanská
*a,
Kateřina
Kolářová
a,
Silvie
Rimpelová
b,
Zdeňka
Kolská
c and
Václav
Švorčík
a
aDepartment of Solid State Engineering, Institute of Chemical Technology, Technická 5, 166 28 Praha 6, Dejvice, Czech Republic. E-mail: vosmansv@vscht.cz; Fax: +420 220 444 330; Tel: +420 220 445 142
bDepartment of Biochemistry and Microbiology, Institute of Chemical Technology Prague, Technická 5, 166 28 Praha 6, Dejvice, Czech Republic. E-mail: rimpelos@vscht.cz; Fax: +420 220 444 330; Tel: +420 220 445 207
cFaculty of Science, J. E. Purkyně University, Moskevská 54, 400 96 Ústí nad Labem, Czech Republic. E-mail: zdenka.kolska@ujep.cz; Fax: +420 220 444 330; Tel: +420 475 2831 44
First published on 4th February 2015
Abstract
The treatment of wounds often deals with bacterial infections which complicate healing. Our aim was to prepare cellulose wound dressings with antibacterial properties. A cellulose dressing was exposed to argon plasma discharge, impregnated with chitosan and then silver chloride particles were precipitated in situ on the dressing's surface. The effect of plasma treatment on both the chitosan impregnation and silver chloride precipitation was studied, together with the antibacterial properties of the prepared dressings. The materials were characterized by optical microscopy, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), goniometry, absorption atomic spectroscopy (AAS) and zeta potential measurement. The antibacterial potency of the dressings was assessed using environmental bacterial strains of Escherichia coli and Staphylococcus epidermidis. Plasma treatment had a positive effect on both chitosan impregnation and the silver chloride precipitation. The antibacterial tests performed on these wound dressings exhibited growth prevention of the two representative strains of Gram-positive and Gram-negative bacteria. These results are of medical interest.
Introduction
Cellulose (β-1,4-glucopyranose) and chitosan (β-1,4-linked 2-acetamido-2-deoxy-D-glucopyranose and β-1,4-linked 2-amino-2-deoxy-D-glucopyranose) are the most abundant naturally occurring polymers. Cellulose wound dressings are commonly used in health care. Interest in the use of chitosan as a pharmaceutical excipient still does not appear to be present in any marketed drugs. However, it is reported as being under investigation for use in a number of pharmaceutical formulations including drug delivery applications such as muco-adhesive and rapid release dosage forms, improved peptide and colonic drug delivery and also systems for gene delivery.1 Nowadays, chitosan is well known polycationic biopolymer with a wide spectrum of biological activities, including antibacterial and antifungal effects.2,3 The antibacterial activity lies in the polycationic character of chitosan. However, it is limited to acidic pH. Chitosan is poorly soluble above pH equal to 6.5, under such conditions deprotonation of amino groups occur and chitosan loses its polycationic character.4,5Cellulose and chitosan structure similarity induces high affinity between both polymers. Cellulose has anionic character and chitosan is readily adsorbed onto fibres by electrostatic interaction.6 More probable intermolecular bonds are based on H-bonds and van der Waals forces. Irreversible adsorption of chitosan onto cellulose is a process predominantly driven by non-electrostatic attraction occurring preferably at low pH, in which its amino groups are charged. For irreversible binding, carboxyl and aldehyde groups present on cellulose are anchoring sites for amino groups of chitosan.7 Ionic or covalent bonds between cellulose and chitosan can be formed under special conditions, one of them is plasma treatment.8
Surface plasma treatment has been used in medicine for modification or sterilization of various materials for a long time. It has been widely used for contact lenses, intraocular lenses, vascular grafts, catheters, filters for blood elements separation and others.9–11 The plasma treatment improves polymer–polymer adhesion.12 More specifically, argon plasma treatment causes ablation and hydrophilization of the treated materials.13 Free radicals are generated during the process which is followed by the cross-linking reactions and reactions of non-saturated species.11 Argon, as an inert gas, is often used to modify and reorganize chemical groups which are already present on the surface.14 This involves breaking old bonds and forming new ones. It was also observed that argon plasma can introduce oxygen onto the surface.15,16
Silver is well-known for its strong antibacterial properties, and is often exploited in topical pharmaceutical products. It is used in the form of Ag nanoparticles, Ag+ ions or in a bound form as a part of a compound. Cytotoxicity issues of the Ag nanoparticles are still somewhat controversial. The Ag+ ions are suspected to be toxic for mammalian cells.17–20 For our wound dressing, silver in the form of insoluble inorganic salt, silver chloride (AgCl), was used. Antimicrobial activity of AgCl can be as significant as the Ag+ ions.21 Silver cations can be bound by chelation on amine groups of chitosan in near neutral solutions. However, the binding mechanism of Ag+ by chitosan is pH-dependent; the amino groups get easily protonated in acidic environment, thus turning the chelation into the electrostatic attraction of anions.22
Since there is a constant need for antibacterial wound dressings, we modified a standard cellulose non-woven wound dressing functionally combining three types of modifications that are considered to be antibacterial – plasma treatment, chitosan impregnation and addition of silver. The aim was to prepare a cellulose-based wound dressing with antibacterial properties, a wound dressing that would not release antimicrobial agens and that might be able to promote healing. The positive effect of chitosan in wound healing promotion was first reported in 1978.23 This work was based on the assumptions that argon plasma treatment oxidizes cellulose, as shown in our previous works24,25 and that the aldehyde and carboxyl groups of cellulose, both original and newly formed after the plasma treatment, would serve as anchoring sites for chitosan.7 The presence of chitosan should then positively influence binding of silver onto the dressing, since chitosan is able to chelate metals and silver has higher attraction to nitrogen than to oxygen.6,26 In another words, one modification step should advantageously influence the next one. Silver in the form of AgCl precipitate was chosen purposely because of the sufficient antibacterial activity and lower toxicity than Ag+ ions.21 Moreover, AgCl is poorly soluble in water, so we supposed to get firmly bonded AgCl precipitate on the wound dressing that would not be released into the aqueous environment.
Materials and methods
Materials
Cellulose dressing, non-woven, perforated, 60 g m−2 was provided from Holzbecher (Zlíč, Czech Republic). Chitosan powder from crab shells, high viscosity, was purchased from Sigma-Aldrich (Steinheim, Germany), AgNO3 from Lachema (Neratovice, Czech Republic), NaCl and acetic acid were purchased from Lach-Ner (Neratovice, Czech Republic). Bacterial strains of environmental Gram-negative Escherichia coli (DBM 3138) and Gram-positive Staphylococcus epidermidis (DBM 3179) were obtained from the Collection of microorganisms of the Institute of Chemical Technology (Prague, Czech Republic). Luria Bertani (LB) broth medium and agar were purchased from Sigma-Aldrich (Steinheim, Germany).Methods
The wound dressing was prepared by three-step modification of non-woven cellulose wound dressing: (i) argon plasma treatment, (ii) impregnation with chitosan, (iii) in situ precipitation of AgCl from silver nitrate (AgNO3) and natrium chloride (NaCl) solutions. Along with the three-step modification, partial modifications were done (not all three steps were performed), samples were compared and the efficiency of each modification step was evaluated. Partial modifications were as follows: cellulose/chitosan, cellulose/plasma/chitosan/, cellulose/AgCl, cellulose/plasma/AgCl and cellulose/chitosan/AgCl. The samples were characterised by optical microscopy, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), goniometry, absorption atomic spectroscopy (AAS) and electrokinetic analysis (zeta potential measurement). Finally, the antibacterial properties were tested using environmental bacterial strains of Escherichia coli and Staphylococcus epidermidis.Plasma treatment
Samples were treated in a direct (glow, diode) Ar+ plasma discharge on Balzers SCD 050 device (BAL-TEC, Lichtenstein) under the following conditions: gas purity 99.997%, flow rate 0.3 dm3 s−1, pressure 10 Pa, electrode distance 50 mm, electrode area 48 cm2, chamber volume approx. 1 dm3, plasma volume 0.24 dm3 and discharge power 5.5 W. Exposure time was 100 s, samples were exposed from both sides. The treatment was accomplished at laboratory conditions (24 °C, 40–60% humidity).27Chitosan impregnation
Cellulose was impregnated with chitosan as follows, analogously to the previously published work of Čakara et al.28: 0.5% (w/v) chitosan solution was prepared. Concentrated acetic acid was used to adjust the pH to 3.6. The solution was heated to 60 °C under vigorous stirring for 1 h. Samples of cellulose (8 × 8 cm2) were placed on Petri dishes and submerged into the prepared solution (50 mL of solution per dish). The dishes were covered with parafilm and left overnight at the room temperature (ca. 15 h). Afterwards the samples were thoroughly rinsed with distilled water, pressed between two layers of filter paper in order to remove the excess of water and left to dry at room temperature.In situ synthesis of AgCl
The in situ synthesis of AgCl particles was performed as follows: the samples were immersed in 0.001 mol dm−3 solution of AgNO3 for 10 min under occasional stirring, followed by immersion in 0.001 mol dm−3 solution of NaCl for 10 min under occasional stirring. The same procedure was repeated twice and the samples were then intensively rinsed in distilled water and left to dry at room temperature. The in situ synthesis was performed similarly to Hu et al. and Klemenčič et al.29,30Material characteristics
Composition of the superficial layer of the samples was studied by X-ray photoelectron spectroscopy (XPS), the spectra were obtained by Omicron Nanotechnology ESCAProbeP spectrometer. The dimension of the exposed area was 2 × 3 mm2. The measurement was done under the following conditions: X-ray source at 1486.7 eV with measuring step 0.05 eV and take-off angle 0° with respect to the surface normal, penetration depth was 1 nm. The measurement was done in ultra-light vacuum. Characteristic carbon (1s), oxygen (1s), nitrogen (1s), silver (3d5/2) and chlorine (2s) peaks were investigated in the spectra, which were evaluated by CasaXPS code software.Surface morphology of the materials and visualization of the AgCl particles was studied by scanning electron microscopy (SEM), device VEGA 3 LMU, TESCAN (Czech Republic), voltage 20 eV. The samples were not sputter coated prior to the measurement. Size of the AgCl particles was evaluated from the SEM images by the NIS-Elements software using statistically significant number of measurements. Optical microscope (LEXT OLS 3100, Olympus, Tokyo, Japan) was used to make images of the cellulose fibres impregnated with chitosan.
Static contact angle experiment was done to visualize the difference in the surface polarity of the samples using conventional goniometry. The measurement was performed at room temperature. Automatic pipette was used to make water drops, 10 μL in volume. The contact angle was calculated by the three point method,31 performed on See System device (Surface Energy Evaluation system, Advex Instruments, Czech Republic).
Electrokinetic analysis was used to examine the changes in chemistry and surface charge at solid–liquid interface. Determination of zeta potential was carried out on SurPASS Instrument (Anton Paar, Austria). Samples were studied in a cylindrical cell in contact with electrolyte (0.001 mol dm−3 KCl) at pH equal to 6.6 and room temperature. The samples were measured four times at constant pH with relative error of 5%. Streaming potential method was used to determine the zeta potential and the Fairbrother–Mastin equation was applied for the calculations.32
For determination of the amount of silver released into the aqueous solution, atomic absorption spectra of the aqueous extracts were recorded. Spectrometer AA880 (Varian Medical Systems, Inc., Palo Alto, CA) with flame atomization was used to determine the total amount of silver in the aqueous extracts. The samples that underwent the precipitation of AgCl were analysed, 1 cm2 of each sample was emerged into 10 ml of deionized water for 24 h and then the extracts were measured. The detection limit of the spectrometer was 0.03 mg dm−3 and the detected amounts of silver in the aqueous extracts were at the edge of the detection limit.
Antibacterial tests
In the final part of the work, antibacterial properties of the modified wound dressings were tested by drip and disc test. Both tests were carried out using E. coli and S. epidermidis bacterial strains. All experiments were accomplished under sterile conditions. To determine the antibacterial activity of the prepared materials, both bacterial cultures were grown overnight in LB broth medium at 37 °C in orbital shaker. Optical densities of the inocula were measured at 600 nm (OD600).For drip test, the cultures were serially diluted with fresh saline solution (PS, 0.9% NaCl, w/v). Samples of the tested materials were cut with a die of 1 cm in diameter. These discs were immersed in 2 ml of PS and inoculated with 1.1 × 104 of colony forming units (CFU) of E. coli and 2.2 × 104 CFU of S. epidermidis. In parallel, E. coli and S. epidermidis incubated only in pure PS were used as controls. The samples were incubated statically at 24 °C for 1 h. Aliquots of 25 μL were dripped on pre-dried LB agar plates. The plates were incubated at 24 °C (E. coli) and 37 °C (S. epidermidis) for 24 h, and then the number of CFU was counted. Each sample was prepared in a triplicate by inoculating three separate discs. Similarly to Kolářová et al.24
Disc test, also called disc diffusion method, consisted in assessment of inhibition zone formed around the samples possessing antibacterial activity, similarly to Bauer et al.33 The overnight bacterial inocula were diluted to achieve final OD600 equal to 2. Bacteria were equally spread on pre-dried LB agar plates. The tested samples, 1 cm in diameter, were placed on the plates with the spread inocula. In parallel, E. coli and S. epidermidis plates without samples were used as positive controls. The plates were incubated statically for 24 h at 24 °C (E. coli) and 37 °C (S. epidermidis). The diameters of the inhibition zones were documented after 24 h growth and then evaluated using ImageJ 1.43 software (NIH, USA). Each sample was prepared in a triplicate using three separate discs.
Results and discussion
Using the three-step modification method described in the previous section, samples of standard non-woven cellulose wound dressing were modified. For schematic illustration of the modifications performed on cellulose (argon plasma treatment, chitosan impregnation and AgCl precipitation) see Fig. 1. Each layer in the scheme represents one modification step, chronologically from the bottom to the top. The results are reported and discussed in the order of the modification steps, antibacterial testing is in the end of this section. | ||
| Fig. 1 Modifications performed on cellulose. Partial modifications: (A) cellulose with precipitated AgCl, (B) cellulose exposed to plasma with precipitated AgCl, (C) cellulose impregnated with chitosan and with precipitated AgCl. Three-step modification: (D) cellulose exposed to plasma, impregnated with chitosan and with precipitated AgCl. | ||
Plasma treatment
The plasma treatment of cellulose led to oxidation of the superficial layer resulting in an increase of polar functional groups content (namely C–O–/C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/O–C–O and OC–O), as shown in detail in our previous work concerning the effect of plasma treatment on cellulose fibre.24 In this work, the effect of argon plasma treatment on non-woven cellulose dressing was examined by the XPS (penetration depth 1 nm) determining the surface composition of the samples, for summarised results see Table 1. Attention was focused on the concentration of carbon (C 1s) and oxygen (O 1s), hydrogen cannot be detected due to the technique limitation. The changes in carbon and oxygen concentration were advantageously expressed by oxygen-to-carbon elemental ratio (O/C ratio). The higher increase of the O/C ratio values the more massive oxidation of the samples. The Table 1 also contains the elemental composition and the O/C ratio of pure cellulose,34 showing a difference of carbon and oxygen content in comparison to pristine cellulose. The O/C ratio of the pristine cellulose sample was 0.33, considerably different from the theoretical value of 0.83, owing to non-cellulosic components (proteins, lignins, pectins, waxes, etc.), which naturally occur in the superficial layer of cotton.35,36 After the plasma treatment of pristine cellulose, the carbon content decreased by 5.7%, meanwhile the oxygen content increased by 5.7% and the increase of the O/C ratio was as high as 0.11. Thus the plasma treatment undoubtedly caused oxidation of cellulose. The same trend was expected and also observed for all of the plasma treated samples. The cellulose/plasma/chitosan samples had the O/C ratio higher by 0.07 than the cellulose/chitosan samples, the cellulose/plasma/AgCl samples had the O/C ratio higher by 0.07 than the cellulose/AgCl samples and finally, the cellulose/plasma/chitosan/AgCl samples had the O/C ratio higher by 0.06 than the cellulose/chitosan/AgCl samples. Clearly, the plasma treatment caused oxidation of the non-woven cellulose dressing and the oxidation was well observed for all the samples exposed to the argon plasma discharge.
O/O–C–O and OC–O), as shown in detail in our previous work concerning the effect of plasma treatment on cellulose fibre.24 In this work, the effect of argon plasma treatment on non-woven cellulose dressing was examined by the XPS (penetration depth 1 nm) determining the surface composition of the samples, for summarised results see Table 1. Attention was focused on the concentration of carbon (C 1s) and oxygen (O 1s), hydrogen cannot be detected due to the technique limitation. The changes in carbon and oxygen concentration were advantageously expressed by oxygen-to-carbon elemental ratio (O/C ratio). The higher increase of the O/C ratio values the more massive oxidation of the samples. The Table 1 also contains the elemental composition and the O/C ratio of pure cellulose,34 showing a difference of carbon and oxygen content in comparison to pristine cellulose. The O/C ratio of the pristine cellulose sample was 0.33, considerably different from the theoretical value of 0.83, owing to non-cellulosic components (proteins, lignins, pectins, waxes, etc.), which naturally occur in the superficial layer of cotton.35,36 After the plasma treatment of pristine cellulose, the carbon content decreased by 5.7%, meanwhile the oxygen content increased by 5.7% and the increase of the O/C ratio was as high as 0.11. Thus the plasma treatment undoubtedly caused oxidation of cellulose. The same trend was expected and also observed for all of the plasma treated samples. The cellulose/plasma/chitosan samples had the O/C ratio higher by 0.07 than the cellulose/chitosan samples, the cellulose/plasma/AgCl samples had the O/C ratio higher by 0.07 than the cellulose/AgCl samples and finally, the cellulose/plasma/chitosan/AgCl samples had the O/C ratio higher by 0.06 than the cellulose/chitosan/AgCl samples. Clearly, the plasma treatment caused oxidation of the non-woven cellulose dressing and the oxidation was well observed for all the samples exposed to the argon plasma discharge.
| Sample | Binding energy (eV) | O/C ratio | ||||
|---|---|---|---|---|---|---|
| 284.7 | 532.4 | 400.1 | 367.7373.7 | 199.6 | ||
| Element composition (at.%) | ||||||
| C (1s) | O (1s) | N (1s) | Ag (3d3/2, 3d5/2) | Cl (2s) | ||
| a Below detection limit. | ||||||
| Pristine cellulose (theoretical) | 54.5 | 45.5 | — | — | — | 0.83 |
| Pristine cellulose | 75.2 | 24.8 | — | — | — | 0.33 |
| Cellulose/plasma | 69.5 | 30.5 | — | — | — | 0.44 |
| Cellulose/chitosan | 69.5 | 29.6 | 0.9 | — | — | 0.46 |
| Cellulose/plasma/chitosan | 64.8 | 34.2 | 1.0 | — | — | 0.53 |
| Cellulose/AgCl | 69.2 | 30.7 | — | 0.05 | a | 0.44 |
| Cellulose/plasma/AgCl | 66.3 | 33.5 | — | 0.09 | 0.08 | 0.51 |
| Cellulose/chitosan/AgCl | 70.5 | 28.5 | 0.7 | 0.11 | 0.11 | 0.40 |
| Cellulose/plasma/chitosan/AgCl | 67.8 | 30.9 | 1.0 | 0.11 | 0.15 | 0.46 |
Chitosan impregnation
After the plasma treatment, the non-woven cellulose dressing was impregnated with chitosan. The presence of chitosan on the sample surface was first investigated by ninhydrin reaction which gives purple colour if the chitosan is present, see Fig. 2.37 Ninhydrin reacts with primary amino groups forming purple colour, so called Ruhemann's purple and the reaction takes 2 min in 100 °C. The primary amino groups of chitosan readily reacted with ninhydrin and the samples turned purple, thus reliably pointing to the presence of chitosan. The pristine cellulose was white (Fig. 2A) and the chitosan impregnated cellulose was purple (Fig. 2B), the fibres were homogeneously covered with chitosan and therefore were purple. The precise amount of chitosan on the sample surface was determined by the XPS, characteristic nitrogen peaks (N 1s) were searched, see Table 1. Nitrogen was found in all samples that were impregnated with chitosan. Otherwise, no nitrogen was found in other samples, which led to the conclusion that nitrogen detected by the XPS in the superficial layer of the samples was originating from chitosan. The cellulose/plasma/chitosan samples had the nitrogen content higher by 0.1 at.% than the cellulose/chitosan samples and the cellulose/plasma/chitosan/AgCl samples had the nitrogen content higher by 0.3 at.%. Such result was expected due to the higher content of the polar functional groups in the plasma treated samples. Concerning the manner of binding of chitosan to cellulose, similarities between cellulose and chitosan enable their high affinity and the intermolecular interactions are very likely based on H-bonds and Van der Waals forces; for irreversible attachment of chitosan to cellulose it is important to introduce carboxyl or aldehyde groups onto the fibres' surface which are then anchoring sites for chitosan.7,38 The polar functional groups acted as nucleophilic binding centres for chitosan that was attracted to cellulose via the positively charged amino groups (the impregnation done under the acidic conditions).39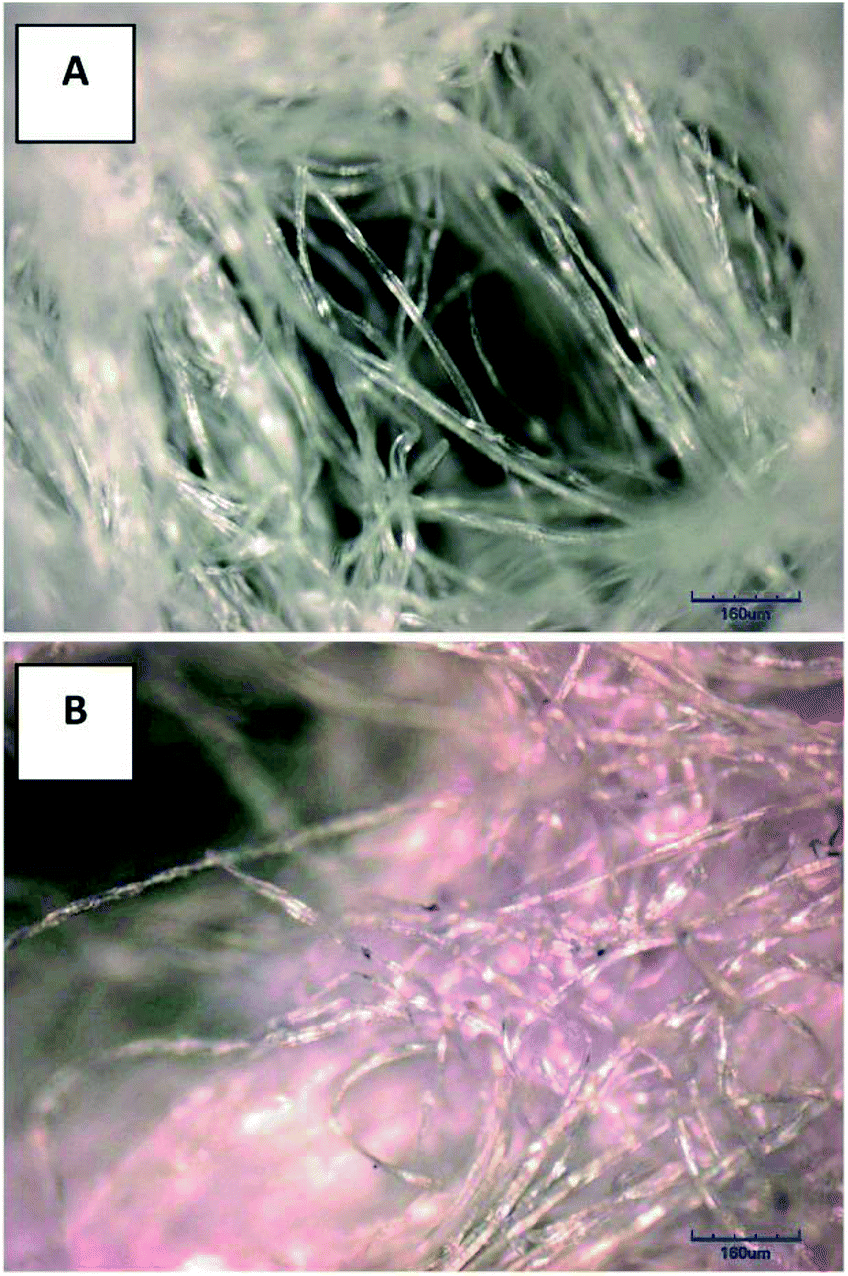 | ||
| Fig. 2 Images of the samples in optical microscope: (A) pristine cellulose, (B) cellulose/chitosan. | ||
As for the reaction mechanism itself, the binding of chitosan to cellulose is presumed to go via the reaction between primary amino groups of chitosan and aldehyde and/or carboxyl groups of cellulose. The reaction of amino group and carboxyl group results in a C–N amide bond40 meanwhile the reaction with aldehyde group leads to the formation of Schiff's base with imine bond.7,41 Pristine cellulose molecule has aldehyde end-groups on C1 and hydroxyl groups on C2, C3 and C6 and no carboxyl groups. During the plasma treatment, the aldehyde groups can undergo oxidation to carboxyl groups and the hydroxyl groups can be oxidized to aldehyde groups or even further to carboxyl groups.40 Argon plasma treatment leads to such oxidation, however the reaction is not selective.24 As was reported before, even the trace amounts of aldehyde groups in cellulose play an important role in a cross-linking to chitosan, which was demonstrated on the composite film formation.41 The composite films derived from the carbonyl-free cellulose had high degree of swelling and low strength in comparison with that from cellulose containing carbonyl groups which was caused by the formation cross-linking composed of Schiff's base bonds and more complex ones originating from the Schiff's base.41
As was stated in the previous section (see plasma treatment), plasma treatment caused increase in the content of C–O–/C–OH and CO/O–C–O and decrease of OC–O functional groups, for details see our previous work,24 and these carboxyl and aldehyde groups were responsible for anchoring of chitosan and the increase of the amount of bounded chitosan on the samples after the plasma treatment. Taking into account the aforementioned, it seems to be likely, that after the plasma treatment chitosan was bound irreversibly on cellulose. It should be noted that cellulose fibres (including wound dressings) are usually weakly acidic due to the pre-treatments such as scouring and bleaching allowing effective chitosan adsorption as well.42
Another phenomenon connected to the chitosan impregnation was the change of polarity of the impregnated samples. The impregnated samples were significantly slowly soaking water in the AgCl precipitation step. Therefore we decided to investigate the wettability of the surface by standard goniometry. Pristine cellulose was hydrophilic (Fig. 3A), the water droplet soaked into the fibres immediately and the contact angle was 0°, meanwhile chitosan impregnated sample was hydrophobic (Fig. 3B) and the water droplet remained on the fibre surface, resulting in a contact angle value of 110 ± 5°. When the samples were treated with plasma and then impregnated with chitosan, they were again hydrophilic and the water droplet soaked into the fibres immediately, the contact angle was 0°. For the cellulose/chitosan/AgCl samples and the cellulose/plasma/chitosan/AgCl samples similar trend was observed. The cellulose/chitosan/AgCl samples absorbed the water droplet immediately, the contact angle was 0°, meanwhile on the cellulose/plasma/chitosan/AgCl samples the water droplet remained, resulting in a contact angle value of 95 ± 5°. Our results show that the plasma pre-treatment positively affected wettability of the chitosan impregnated samples.
 | ||
| Fig. 3 Images of the water droplet on the surface of (A) cellulose/plasma/chitosan and (B) cellulose/chitosan surface. | ||
The difference in the surface wettability was closely connected to the way of binding of chitosan to cellulose. In the work of Hosokawa et al.41 was documented that a composite film of chitosan and carbonyl-free cellulose had a high swelling degree which was not affected with the increase of carboxyl groups. On the other hand, the swelling degree decreased with the increasing content of carbonyl groups in cellulose. In conclusion, the reduction of swelling degree enhances the cross-linking structures in the film. These cross-linkings are composed of Shiff's base bonds and more complex ones originating from Schiff's base.41 Based on these findings, the low surface wettability of the cellulose/chitosan (110 ± 5°) and cellulose/chitosan/AgCl (95 ± 5°) samples was caused by the binding of chitosan to the aldehyde groups via Schiff's base and the following formation of the cross-linkings. This was further supported by the XPS analysis of pristine and plasma treated cellulose. Deconvolution of the carbon peak showed that the amount of CO/O–C–O groups decreased significantly after the plasma treatment; from 40.7 at.% to 21.8 at.%. On the contrary, the amount of the OC–O groups increased after the plasma treatment, from 5.9 at.% to 9.9 at.%. Such high content of aldehyde groups in the pristine cellulose and low content of carboxyl groups ensured cross-linking with chitosan and thus low surface wettability. The plasma treated samples did not have such amount of binding sites for the formation of Schiff's base and cross-linkings so the resulting wettability was thus affected by the formation of amide bonds.
The overall water absorption estimated by gravimetry was not comprised in this study, since we studied that in detail our previous work.24
To support the above mentioned, the changes introduced by the plasma treatment and chitosan impregnation were studied by the zeta potential measurement. The electrokinetic potential (zeta potential) at the interface of sample surface and liquid is caused, among others, by ionization or dissociation of surface functional groups.43 The negative charge of the cellulose samples resulted from the adsorption of chloride and hydroxide ions from the KCl water solution during the measurement.44 The positive charge (meaning the positive increment to the overall zeta potential) was caused by the presence of chitosan amino groups that were ionized (–NH3+) and therefore the surface was less negatively (more positively) charged than cellulose without impregnation. Furthermore, the dissociation of acidic or basic groups during the zeta potential measurement is considered to be equivalent to the adsorption of hydroxide or hydroxonium ions.45
In the case of the chitosan impregnated samples, they showed less negatively (more positively) charged surface than pristine cellulose (Table 2). The highest zeta potential, −4.0 mV, was observed for samples treated by plasma and then impregnated with chitosan. Pristine cellulose had the zeta potential of −18.0 mV, meanwhile the zeta potential of plasma treated cellulose was more negative, which was caused by the oxidation documented by the XPS measurement (Table 1). The difference between the cellulose/plasma/chitosan samples and the cellulose/chitosan samples was interesting, the higher zeta potential of the cellulose/plasma/chitosan samples depended on the amount of positively charged groups on the surface. Therefore we assumed that there was more chitosan bound on the surface of the samples that were treated by plasma discharge prior to the impregnation. The XPS results confirmed our hypothesis. In addition, Zemljič et al. published that the oxygen plasma treatment of cellulose improved adsorption of chitosan onto cellulosic fabric, which is consistent with our results.39,42
| Sample | Zeta potential (mV) |
|---|---|
| Pristine cellulose | −18.0 |
| Cellulose/plasma | −25.0 |
| Cellulose/chitosan | −8.5 |
| Cellulose/plasma/chitosan | −4.0 |
AgCl precipitation
AgCl particles were formed on the sample's surface via in situ reaction in AgNO3 and NaCl solutions. The particles were visible in the SEM images (Fig. 4) and they were spread homogenously on the surface, the amount and size varied. The cellulose/AgCl samples (Fig. 4A) had low amount of AgCl particles, on the other hand the three-step-modified samples (Fig. 4D) had a significant amount of AgCl particles, according to the XPS (Table 1) it was twice more. We found out that the amount of silver on the samples was rising in this order: pristine cellulose/AgCl < cellulose/plasma/AgCl < cellulose/chitosan/AgCl < cellulose/plasma/chitosan/AgCl. The size of the AgCl particles on the samples was also investigated. It varied from 256 to 501 nm, the size of AgCl particles was rising in the order: pristine cellulose/AgCl < cellulose/plasma/AgCl < cellulose/chitosan/AgCl < cellulose/plasma/chitosan/AgCl, showing the identical trend as the amount of silver on the samples. AgCl particles on the cellulose/AgCl sample had 256 nm, cellulose/plasma/AgCl 283 nm, cellulose/chitosan/AgCl 417 nm and cellulose/plasma/chitosan/AgCl 501 nm. Compared to the work of Hu et al.,29 the time factor of the precipitation process played a crucial role in the size of the AgCl particles. They immersed samples of bacterial cellulose for 1 min into AgNO3 and for 1 min into NaCl solution, resulting in AgCl formation with the mean size of 25.8 nm. The immersion time in our work was 10× longer and the particles were approximately 10× bigger. | ||
| Fig. 4 SEM images of the samples with in situ prepared AgCl particles. (A) cellulose/AgCl, (B) cellulose/plasma/AgCl, (C) cellulose/chitosan/AgCl and (D) cellulose/plasma/chitosan/AgCl. | ||
Silver has high affinity for nitrogen, lower for oxygen and much lower for carbon.26 Nitrogen atoms have free electron doublets that are able to react with silver cations, because they have lone electron pairs in orbitals. Oxygen is able to attract the silver via electrostatic interactions, but the attraction is weaker than in the case of nitrogen.28,29 In the case of cellulose, silver ions are attracted to hydroxyl and ether groups. In the case of chitosan, amine groups in chitosan are strongly attractive for silver cations. In another words, the affinity of silver to nitrogen prevailed over the affinity to oxygen. However, it must be pointed out that the uptake mechanism of silver cations by amine groups is pH-dependent; the amine groups get easily protonated (–NH3+) in acidic environment, thus turning the chelation of cations into the electrostatic attraction of anions.22 Therefore it was important to rinse the chitosan impregnated samples in distilled water because the impregnation was done under acidic conditions.
Comparing the cellulose/AgCl and cellulose/plasma/AgCl samples, more silver was found in the plasma treated samples. This was related to the fact that plasma exposure led to oxidation, more oxygen functional groups were situated on the sample surface and this oxygen attracted the silver. In the case of cellulose/AgCl and cellulose/chitosan/AgCl, up to twice more silver was found in the cellulose/chitosan/AgCl samples, for the result see Table 1. The difference was caused by the presence of nitrogen in chitosan that attracted and chelated the silver.
We also wondered whether the silver was irreversibly bounded on the wound dressing or not, therefore we performed a leaking test. An aqueous extracts of the samples with AgCl were analyzed and the AAS results showed that leaking of AgCl from the samples was considerably low, owing to the fact that AgCl is poorly soluble in water. AgCl had to be securely bounded on the samples. Results from the AAS were 0.054 mg l−1 for cellulose/AgCl, 0.031 mg l−1 for cellulose/plasma/AgCl, 0.056 mg l−1 for cellulose/chitosan/AgCl and 0.075 mg l−1 for cellulose/plasma/chitosan/AgCl. The standard error of the AAS measurement was 0–10% and the detection limit for silver was 0.03 mg l−1. The results were only slightly above the detection limit and on the edge of the standard error of the measurement, thus concluding that the release of silver from the samples to the aqueous environment was negligible.
Antibacterial test
At last, samples were submitted to antibacterial test to investigate their potency against E. coli and S. epidermidis. Two different assays were performed: drip test and disc test. The drip test showed 100% reduction of E. coli within 24 h for all samples containing silver (Fig. 5). Clearly, the AgCl particles were responsible for the antibacterial effect on E. coli. The cellulose/chitosan samples showed confluent growth so the E. coli CFU were not countable due to the lower bactericidal activity of chitosan on Gram-negative bacteria strains.46 The number of CFU of E. coli grown on the cellulose/plasma/chitosan samples were comparable to the control (bacterial cells incubated only in the physiological solution of 0.9% NaCl), thus suggesting that the plasma treatment itself had no preventing effect on the growth of E. coli. | ||
| Fig. 5 Drip test of antibacterial properties of the prepared samples on E. coli. | ||
For the results of the drip test performed on S. epidermidis see Fig. 6. The results were particularly interesting, since the growth of S. epidermidis was not completely reduced and the difference between the samples could be clearly observed. Generally, the growth of S. epidermidis was diminished from 50 up to 75% for all samples within 24 h, in comparison to the control. More specifically, the samples containing silver showed better reduction of the growth than the samples without silver, however the cellulose/chitosan and cellulose/plasma/chitosan samples showed sufficiently high growth reduction as well, up to 50%. Concerning the antibacterial effect of chitosan on S. epidermidis, the data were consistent with the literature.2,3 When the samples were arranged in pairs, interesting results were observed. The cellulose/chitosan and cellulose/plasma/chitosan samples showed very similar result, the growth reduction of S. epidermidis being a little bit lower for the samples that were treated by argon plasma prior to the chitosan impregnation. This was in agreement with our prior study that plasma treated cellulose underwent structure degradation and slightly supported the bacterial growth on the samples.24 This could be well observed for the pair cellulose/AgCl and cellulose/plasma/AgCl, where the difference in the bacterial growth is more pronounced and even better observed for the pair cellulose/plasma/chitosan and cellulose/plasma/AgCl that showed that plasma pre-treatment lowered the antibacterial efficiency of AgCl if compared to the pair cellulose/chitosan and cellulose/AgCl.
 | ||
| Fig. 6 Drip test of antibacterial properties of prepared samples on S. epidermidis. | ||
As regards the efficiency of antibacterial properties, it is evident that AgCl reduced the bacterial growth more than chitosan, see the bars in Fig. 6. However, the situation gets interesting when it comes to the combination of both chitosan and AgCl. When the cellulose/chitosan, cellulose/AgCl samples and cellulose/chitosan/AgCl samples were compared, it was evident that there was a trend of rising efficiency against bacterial growth. The same trend was observed in the order of the cellulose/plasma/chitosan, cellulose/plasma/AgCl and cellulose/plasma/chitosan/AgCl samples. Based on these results, the combination of chitosan and AgCl treatment increased the antibacterial efficiency against S. epidermidis. As regards the meaning of plasma treatment, the cellulose/chitosan/AgCl samples showed the highest reduction of the bacterial growth, but their surface exhibited impaired wettability (Fig. 3), whereas with the use of plasma pre-treatment the cellulose/plasma/chitosan/AgCl samples exhibited good wettability and kept very good antibacterial activity.
When comparing the drip test for E. coli and S. epidermidis, our results were consistent with the work of Xia et al., that chitosan generally showed stronger bactericidal effect on Gram-positive bacteria than Gram-negative bacteria.3
In the disc test, inhibition zones were observed around the samples with AgCl and no inhibition zones around other samples. For typical inhibition zone see Fig. 7 and for the summarized data see Table 3. The best results were observed for the cellulose/plasma/chitosan/AgCl samples, which is also consistent with the XPS results (Table 1). The highest amount of antibacterially active silver was detected in these samples. Even though the area of the zones of inhibition was not as big as was expected, it was still very good, most probably owing to the poor solubility of AgCl in water, thus not diffusing to the LB-agar medium. The plasma treated samples showed better results than the non-treated ones; the inhibition zone of cellulose/plasma/AgCl was larger than for cellulose/AgCl and the same trend was observed for cellulose/plasma/chitosan/AgCl and cellulose/chitosan/AgCl. The antibacterial effect of the samples impregnated only with chitosan (and no AgCl) was not observed, although it should have resulted in the elimination of the bacterial growth.46,47 The most probable explanation is that the concentration of chitosan on the samples' surface was not sufficient for the disc test and did not reach the minimum inhibitory concentration,47,48 even though the antibacterial activity was first proven in the drip test (Fig. 5 and 6). For both bacterial strains, E. coli and S. epidermidis, was experienced the same result.
 | ||
| Fig. 7 The zone of inhibition (clear ring around the sample) induced by the cellulose/plasma/chitosan/AgCl sample on E. coli. | ||
| Sample | Zone of inhibition (cm) | Radius (cm) | Area (cm2) |
|---|---|---|---|
| Cellulose/chitosan | 0 | 0 | 0 |
| Cellulose/plasma/chitosan | 0 | 0 | 0 |
| cellulose/AgCl | 0.13 | 0.63 | 1.25 |
| Cellulose/plasma/AgCl | 0.17 | 0.67 | 1.41 |
| Cellulose/chitosan/AgCl | 0.20 | 0.70 | 1.54 |
| Cellulose/plasma/chitosan/AgCl | 0.22 | 0.72 | 1.63 |
Conclusion
The three-step modification of the standard cellulose wound dressing presented in this study is cost-effective, nature-friendly and does not require large and complicated device. The wound dressing modified by all the three steps (argon plasma treatment, chitosan impregnation and AgCl precipitation) had the best outcome. It possessed antibacterial activity against E. coli and S. epidermidis, the surface of the dressing was hydrophilic and the highest amounts of chitosan and AgCl were found in these samples. In order to evaluate the impact of plasma treatment, it oxidized the surface which led to the higher amount of chitosan impregnated onto the surface and to the increase of surface hydrophilicity. Apart from improved chitosan impregnation, the plasma treatment almost doubled the amount of AgCl precipitated on the plasma activated surface. Positive influence of one modification step on the next was proved in our work. Chronologically, argon plasma treatment positively affected the chitosan impregnation and more chitosan was adsorbed. Chitosan was used here due to the antibacterial and healing promoting activity. The adsorbed chitosan then positively affected the amount of the AgCl in the superficial layer of the sample and finally, AgCl along with chitosan were responsible for the antibacterial effect. Interesting result was that the combination of chitosan impregnation and AgCl precipitation showed better antibacterial effect than each of them alone. Therefore, to the best of our knowledge this three-step method can be recommended for preparation of the antibacterial wound dressings.Acknowledgements
This work was financially supported by GA CR under the project no. P108/12/1168.Notes and references
- P. Baldrick, Regul. Toxicol. Pharmacol., 2010, 56, 290–299 CrossRef CAS PubMed.
- R. Jayakumar, D. Menon, K. Manzoor, S. V. Nair and H. Tamura, Carbohydr. Polym., 2010, 82, 227–232 CrossRef CAS.
- W. Xia, P. Liu, J. Zhang and J. Chen, Food Hydrocolloids, 2011, 25, 170–179 CrossRef CAS.
- X. Fei Liu, Y. Lin Guan, D. Zhi Yang, Z. Li and K. De Yao, J. Appl. Polym. Sci., 2001, 79, 1324–1335 CrossRef.
- S.-H. Lim and S. M. Hudson, Carbohydr. Res., 2004, 339, 313–319 CrossRef CAS PubMed.
- P. Myllytie, J. Salmi and J. Laine, BioResources, 2009, 4, 1647–1662 CAS.
- S. Strnad, O. Sauperl and L. Fras Zemljič, in Biopolymers, ed. M. Elnashar, InTech, 2010 Search PubMed.
- A. Vesel, M. Mozetic, S. Strnad, Z. Peršin, K. Stana-Kleinschek and N. Hauptman, Vacuum, 2009, 84, 79–82 CrossRef CAS.
- P. K. Chu, J. Y. Chen, L. P. Wang and N. Huang, Mater. Sci. Eng., R, 2002, 36, 143–206 CrossRef.
- S. M. Luna, S. S. Silva, M. E. Gomes, J. F. Mano and R. L. Reis, J. Biomater. Appl., 2011, 26, 101–116 CrossRef CAS PubMed.
- A. J. Moreira, R. D. Mansano, T. d. J. Andreoli Pinto, R. Ruas, L. d. S. Zambon, M. V. da Silva and P. B. Verdonck, Appl. Surf. Sci., 2004, 235, 151–155 CrossRef CAS.
- D. T. Clark and A. Dilks, J. Polym. Sci., Part A: Polym. Chem., 1979, 17, 957–976 CrossRef CAS.
- J. Siegel, A. Reznickova, A. Chaloupka, P. Slepicka and V. Svorcik, Radiat. Eff. Defects Solids, 2008, 163, 779–788 CrossRef CAS.
- V. Švorčík, K. Kolářová, P. Slepička, A. Macková, M. Novotná and V. Hnatowicz, Polym. Degrad. Stab., 2006, 91, 1219–1225 CrossRef.
- P. Slepička, S. Trostová, N. Slepičková Kasálková, Z. Kolská, P. Sajdl and V. Švorčík, Plasma Processes Polym., 2012, 9, 197–206 CrossRef.
- Z. Zhou, J. Wang, X. Huang, L. Zhang, S. Moyo, S. Sun and Y. Qiu, Appl. Surf. Sci., 2012, 258, 4411–4416 CrossRef CAS.
- M. Ahamed, M. Karns, M. Goodson, J. Rowe, S. M. Hussain, J. J. Schlager and Y. Hong, Toxicol. Appl. Pharmacol., 2008, 233, 404–410 CrossRef CAS PubMed.
- K. Kawata, M. Osawa and S. Okabe, Environ. Sci. Technol., 2009, 43, 6046–6051 CrossRef CAS PubMed.
- S. Prabhu and E. Poulose, Int. Nano Lett., 2012, 2, 1–10 CrossRef.
- J. Siegel, K. Kolářová, V. Vosmanská, S. Rimpelová, J. Leitner and V. Švorčík, Mater. Lett., 2013, 113, 59–62 CrossRef CAS.
- O. Choi, K. K. Deng, N. J. Kim, L. Ross Jr, R. Y. Surampalli and Z. Hu, Water Res., 2008, 42, 3066–3074 CrossRef CAS PubMed.
- E. Guibal, Sep. Purif. Technol., 2004, 38, 43–74 CrossRef CAS.
- L. Ilium, Pharm. Res., 1998, 15, 1326–1331 CrossRef.
- K. Kolarova, V. Vosmanska, S. Rimpelova and V. Svorcik, Cellulose, 2013, 20, 953–961 CrossRef CAS.
- V. Vosmanska, K. Kolarova, S. Rimpelova and V. Svorcik, Cellulose, 2014, 21, 2445–2456 CrossRef CAS.
- S. S. Grazhulene, A. N. Red'kin, G. F. Telegin, A. V. Bazhenov and T. N. Fursova, J. Anal. Chem., 2010, 65, 682–689 CrossRef CAS.
- V. Švorčík, A. Řezníčková, P. Sajdl, Z. Kolská, Z. Makajová and P. Slepička, J. Mater. Sci., 2011, 46, 7917–7922 CrossRef.
- D. Čakara, L. Fras, M. Bračič and K. S. Kleinschek, Carbohydr. Polym., 2009, 78, 36–40 CrossRef.
- W. Hu, S. Chen, X. Li, S. Shi, W. Shen, X. Zhang and H. Wang, Mater. Sci. Eng., C, 2009, 29, 1216–1219 CrossRef CAS.
- D. Klemenčič, B. Tomšič, F. Kovač and B. Simončič, Cellulose, 2012, 19, 1715–1729 CrossRef.
- A. Marmur, Soft Matter, 2006, 2, 12–17 RSC.
- Z. Kolská, A. Řezníčková and V. Švorčík, e-Polymers, 2012, 12, 960–972 CrossRef.
- A. W. Bauer, W. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol., 1966, 45, 496–496 Search PubMed.
- T. Topalovic, V. A. Nierstrasz, L. Bautista, D. Jocic, A. Navarro and M. M. C. G. Warmoeskerken, Colloids Surf., A, 2007, 296, 76–85 CrossRef CAS.
- A. Calvimontes, P. Mauersberger, M. Nitschke, V. Dutschk and F. Simon, Cellulose, 2011, 18, 803–809 CrossRef CAS.
- H. A. Karahan and E. Özdoğan, Fibers Polym., 2008, 9, 21–26 CrossRef CAS.
- R. Jelly, S. W. Lewis, C. Lennard, K. F. Lim and J. Almog, Talanta, 2010, 82, 1717–1724 CrossRef CAS PubMed.
- A. L. Da Róz, F. L. Leite, L. V. Pereiro, P. A. P. Nascente, V. Zucolotto, O. N. Oliveira and A. J. F. Carvalho, Carbohydr. Polym., 2010, 80, 65–70 CrossRef.
- L. F. Zemljič, Z. Peršin, P. Stenius and K. S. Kleinschek, Cellulose, 2008, 15, 681–690 CrossRef.
- Y. Xu, C. Qiu, X. Zhang and W. Zhang, Carbohydr. Polym., 2014, 112, 186–194 CrossRef CAS PubMed.
- J. Hosokawa, M. Nishiyama, K. Yoshihara, T. Kubo and A. Terabe, Ind. Eng. Chem. Res., 1991, 30, 788–792 CrossRef CAS.
- L. Zemljič Fras, Z. Peršin and P. Stenius, Biomacromolecules, 2009, 10, 1181–1187 CrossRef PubMed.
- Z. Kolska, Z. Makajova, K. Kolarova, N. Kasalkova, S. Trostova, A. Reznickova, J. Siegel and V. Svorcik, in Polymer Science, ed. F. Yilmaz, InTech, 2013 Search PubMed.
- M. Elimelech, W. H. Chen and J. J. Waypa, Desalination, 1994, 95, 269–286 CrossRef CAS.
- K. Stana-Kleinschek, S. Strnad and V. Ribitsch, Polym. Eng. Sci., 1999, 39, 1412–1424 CAS.
- J.-S. Kim and Y. Kim, Nutr. Res. Pract., 2007, 1, 273–278 CrossRef CAS PubMed.
- H. K. No, N. Young Park, S. Ho Lee and S. P. Meyers, Int. J. Food Microbiol., 2002, 74, 65–72 CrossRef CAS PubMed.
- L.-Y. Zheng and J.-F. Zhu, Carbohydr. Polym., 2003, 54, 527–530 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |