
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Positively charged, surfactant-free gold nanoparticles for nucleic acid delivery†
Jianfeng
Guo
a,
Mark J.
Armstrong
bc,
Caitriona M.
O'Driscoll
a,
Justin D.
Holmes
bc and
Kamil
Rahme
*bcd
aPharmacodelivery group, School of Pharmacy, University College Cork, Cork, Ireland
bMaterials Chemistry and Analysis Group, Department of Chemistry and the Tyndall National Institute, University College Cork, Cork, Ireland
cCentre for Research on Adaptive Nanostructures and Nanodevices (CRANN), Trinity College Dublin, Dublin 2, Ireland
dDepartment of Sciences, Faculty of Natural and Applied Science, Notre Dame University (Louaize), Zouk Mosbeh, Lebanon. E-mail: kamil.rahme@ndu.edu.lb; Fax: +961 9 225164; Tel: +961 9 218950
First published on 3rd February 2015
Abstract
Positively charged, surfactant–free gold nanoparticles (Au NPs) with diameters ranging between 2–200 nm have been synthesised in water via a seed-mediated growth method, involving the reduction of gold(III) chloride (AuCl3) by hydroxylamine hydrochloride (NH2OH·HCl) in the presence of L-cysteine methyl ester hydrochloride (HSCH2CH(NH2)COOCH3·HCl) as a capping agent. The mercapto group (–SH) on the capping ligand has a high affinity for Au, anchoring the cysteine group to the nanoparticles, whilst the ammonium group (–NH3+), formed by the presence of an amine group in slightly acidic media (pH ∼ 4.5–5), resulted in positively charged colloidal nanoparticles (ζ-potential +33 to +49 mV), which was key to their electrostatic stability. Data from cytotoxicity studies performed on a range of different cell types (human and murine), including human prostate cancer cells (PC3), showed that the positively charged Au–L-cysteine–cysteine nanoparticles were less cytotoxic than positively charged Au NPs produced using commonly employed surfactant cetyl trimethyl ammonium bromide (CTAB) under similar conditions. In addition, the positively charged Au NPs could be successfully complexed with small interfering RNA (siRNA). At the cellular level, the uptake of fluorescein siRNA from the charged nanoparticles was comparable to uptake from the commercial carrier INTERFRin™, implying the potential application of these novel vectors for nucleic acid delivery.
1. Introduction
Gold nanoparticles (Au NPs) due to their unique physical, chemical and optical properties have been widely used in biomedical applications such as biosensors, clinical chemistry, immunoassays, optical imaging and the monitoring of cells and tissues.1–6 As the gold core is generally inert, toxic-free and biocompatible, it is a favourable starting material for carrier construction.7–13 For instance, positively charged gold nanoparticles have been developed for drug and gene delivery.14–18 In general, cationic gold nanoparticles are synthesised in the presence of surfactants such as cetyl trimethyl ammonium bromide (CTAB).19–21 Of note, positively charged Au nanoprisms have been synthesised by Millstone et al. using a three-step mediated seed growth process in an aqueous solution of CTAB at a concentration of 50 mM (50 times the critical micelle concentration of CTAB).22 In addition, Au nanostructures with shapes ranging from 3D octahedra, triangles, to 2D hexagonal nanoplates have been obtained through changes in the CTAB concentration, used as both a reducing agent and capping ligand during hydrothermal treatment of chloroauric acid.23The successful application of Au nanoparticles in biomedical applications requires tunable particle sizes and morphologies as well as an assessment of their toxicity.24–28 The presence of surfactants during the formation of Au nanoparticles may affect their final size and their shape diversity; this is due to specific adsorption of Au–surfactant complexes on the facets of the nanoparticle seeds that limit their growth and enhance their anisotropic shape.21,29,30 In addition, many studies have shown that the biological uptake of positively charged nanoparticles is higher than their negatively charged counterparts, but with positively charged Au nanoparticles produced from CTAB showing a high level of biological toxicity.31–33 Therefore, in the synthesis of Au nanostructures for biological applications, there is a requirement to avoid the use of highly toxic chemicals and surfactants such as CTAB, and move towards surfactant-free synthesis methods.34,35
Alternatively, Au NPs using various cationic materials as protecting agents, e.g. amine, polyethylenimine (PEI), poly-L-lysine (PLL), poly (diallyl dimethylammonium) chloride (PDDA), cationic chitosan and lipids, have recently been developed to produce positively charged Au NPs for application in the delivery of biomolecules.14–18,36–38 For instance, Bhumkar and co-workers reported the transmucosal delivery of insulin using chitosan-reduced Au NPs.15 In this study, chitosan was used as a reducing agent to produce the Au NPs, and being a polyelectrolyte it also played a role as an electrostatic stabiliser. The oral and nasal administration of insulin formulated with chitosan-reduced Au NPs improved the pharmacodynamics efficiency in terms of blood glucose levels (BGL) in diabetic rats.15 Furthermore, Kong et al. developed cationic lipid-coated Au NPs (L-Au NPs) for the efficient delivery of therapeutic siRNA,16 In Kong's study, hydrophobic dodecanethiol-capped Au NPs nanoparticles of about 5 nm were produced through the reduction of a Au(PPh3)Cl complex in presence of dodecanethiol using Borane-tert-butylamine complex as reducing agent in benzene, a method well known to produce gold nanoparticles with diameters below 10 nm. These nanoparticles were transferred into water by using a modified emulsification/solvent evaporation method.16 Three different lipid components L-α-dioleoyl phosphatidylethanolamine (DOPE), cholesterol hydrochloride (Chol) and (3β-[N-(N′,N′-dimethylaminoethane)-carbamoyl]-cholesterol (DC-Chol) were used to coat the hydrophobic dodecanethiol-capped Au NPs through hydrophobic–hydrophobic interactions. The resultant cationic lipid-coated gold nanoparticles (L-Au NPs) were obtained after several purifications through centrifugation and excessive dialysis with deionised water, after which they were also able to effectively condense siRNA through electrostatic interactions and demonstrated efficient gene knockdown in a variety of cell lines without showing significant cytotoxicity.16
In this article we describe a seeded-growth method for synthesising nearly spherical positively charged Au nanoparticles directly in water over a wide range of mean diameters (2–200 nm), avoiding the use of toxic surfactants and organic solvents such as benzene or toluene. L-Cysteine methyl ester hydrochloride (HSCH2CH(NH2)COOCH3·HCl) was chosen as the capping agent, because the mercapto group (–SH) is known to have a high affinity for Au, allowing anchoring of cysteine to the nanoparticles,39,40 while the ammonium group (–NH3+), formed by the presence of an amine group in slightly acid aqueous media, results in positively charged nanoparticles. The presence of the positive charges on the surface of Au NPs is a key to the electrostatic stability of the final aqueous colloidal solution. Additionally, cytotoxicity studies of positively charged Au–L-cysteine nanoparticles were performed using human and murine cell lines, showing improved cell viability compared to positively charged and excessively purified Au NPs synthesised using CTAB as a capping ligand. Moreover, Au NPs with diameters greater than 100 nm demonstrated effective complexation of siRNA (one of RNA interference (RNAi) effectors, a class of double-stranded RNA molecules, which can regulate gene expression post-transcriptionally). The Au NPs demonstrated comparable cellular uptake of siRNA relative to INTERFRin™ (a PEI-based commercial transfection reagent), suggesting their role as a promising nucleic acid delivery vector.
2. Experimental
2.1. Chemicals and materials
Gold(III) chloride (AuCl3), sodium borohydride (NaBH4), hydroxylamine hydrochloride (NH2OH·HCl), cetyl trimethyl ammonium bromide (Au NPs–CTAB) were purchased from Sigma Aldrich. L-Cysteine methyl ester hydrochloride (HSCH2CH(NH2)COOCH3·HCl) was obtained from Fluka. All products were used as received. Purified H2O (resistivity ≈ 18.2 MΩ cm) was used as a solvent. All glassware was cleaned with aqua regia (3 parts of concentrated HCl and 1 part of concentrated HNO3), rinsed with distilled water, ethanol, and acetone and oven-dried before use.2.2. Preparation of Au NPs
For example by adding 40 mL of the colloidal Au NPs–L-cysteine seeds solution prepared above, a colour change of the solution from brown to brown slightly reddish was observed. The resultant nanoparticles obtained had a mean diameter of 4 ± 1.5 nm as determined from TEM images analysis and labeled GR1, decreasing volume of the colloidal Au NPs–L-cysteine seeds solution from 40 mL to 25 mL, resulted nanoparticles with mean diameter of 5.5 ± 2 nm as determined from TEM images analysis and labeled GR2. In order to avoid increasing the polydispersity of the samples a successive growth was used, therefore, for GR3 Au NPs–L-cysteine diameter of 7 ± 2.5 nm as determined from TEM images analysis, 20 mL of GR2 Au NPs–L-cysteine solution prepared above were used as seeds and the solution was stirred for approximately 4 h. For the rest of the samples the GR4-G12 the size was determined using SEM. For GR4 Au NPs–L-cysteine diameter of 9 ± 4 nm 20 mL of GR3 Au NPs–L-cysteine were used as seeds (added to 40 mL AuCl3 0.5 mmol L−1). For GR5 Au NPs–L-cysteine diameter of 16 ± 2 nm, 10 mL of GR4 Au NPs–L-cysteine solution were used as seeds. For GR6 Au NPs–L-cysteine diameter of 46 ± 4 nm, 10 mL of GR5 Au NPs–L-cysteine solution were used as seeds. For GR7 Au NPs–L-cysteine diameter of 56 ± 4 nm, 5 mL of GR5 Au NPs–L-cysteine seeds solution were used. For GR8 Au NPs–L-cysteine diameter of 80 ± 6 nm, 5 mL of GR6 Au NPs–L-cysteine solution were used as seeds. For GR9 Au NPs–L-cysteine diameter of 104 ± 7 nm, 5 mL of GR7 Au NPs–L-cysteine solution were used as seeds. For GR10 Au NPs–L-cysteine diameter of 118 ± 9 nm: 1.5 mL of GR7 Au NPs–L-cysteine were used as seeds (added to 28.3 mL of AuCl3 0.5 mmol L−1). For GR11 (145 ± 18 nm) and GR 12 (195 ± 20 nm) Au NPs–L-cysteine, 5 mL and 1 mL of GR10 Au NPs–L-cysteine solution used as seeds respectively (added to 28.3 mL of AuCl3 0.5 mmol L−1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and redispersion in water.43 Solutions of Au NPs–CTAB with mean diameters around 60 nm were synthesised in a similar method used for the Au NPs–cysteine solutions, with the only difference that the 20 nm Au NPs–CTAB solutions were used as seeds. Briefly, to an aqueous solution (40 mL) of AuCl3 (0.6 mmol L−1) was added 14 mL of 20 nm Au NPs–CTAB and L-cysteine methyl ester hydrochloride (0.5 mmol L−1) the mixture was stirred gently afterward 0.33 mL of 115.8 mmol L−1 NH2OH, HCl was added and the solution was stirred for approximately 18 h. The resultant nanoparticles obtained had a mean diameter of 60 nm (σ = 8 nm).
000 rpm) and redispersion in water.43 Solutions of Au NPs–CTAB with mean diameters around 60 nm were synthesised in a similar method used for the Au NPs–cysteine solutions, with the only difference that the 20 nm Au NPs–CTAB solutions were used as seeds. Briefly, to an aqueous solution (40 mL) of AuCl3 (0.6 mmol L−1) was added 14 mL of 20 nm Au NPs–CTAB and L-cysteine methyl ester hydrochloride (0.5 mmol L−1) the mixture was stirred gently afterward 0.33 mL of 115.8 mmol L−1 NH2OH, HCl was added and the solution was stirred for approximately 18 h. The resultant nanoparticles obtained had a mean diameter of 60 nm (σ = 8 nm).
2.3. UV-visible spectroscopy
Optical absorption spectra were obtained on a CARY UV-visible spectrophotometer with a Xenon lamp (300–900 nm range, 0.5 nm resolution).2.4. Particle size and zeta potential measurements
The pristine solutions of Au NPs were measured by dynamic light scattering (DLS). The measurements were undertaken with a Malvern instrument (Zeta sizer Nano Series) at 25 °C using the default non-invasive back scattering (NIBS) technique with a detection angle of 173°. Three measurements were made per sample and the standard deviation (σ) was calculated, typically σ = 1–2 nm.2.5. Transmission electron microscopy
Au nanoparticles were placed on carbon-coated copper grids (Quantifoil, Germany) and dried over night in air, prior to transmission electron microscope (TEM) inspection. The samples were inspected using a JEOL JEM-2100 TEM operating at 200 kV. Micrographs were recorded on a Gatan 1.35 K × 1.04 K × 12 bit ES500W CCD camera. TEM images were analysed using Image J software.2.6. Scanning electron microscopy
Au nanoparticles were deposited from solution onto a Si wafer and dried in air prior to inspection by scanning electron microscope (SEM). The samples were inspected using a FEI 630 NanoSEM equipped with an Oxford INCA energy dispersive X-ray (EDX) detector, operating at 5 kV.2.7. Cell culture and cytotoxicity
Hep G2, Caco-2, PC-3, B16F10 and CT26 cell lines were purchased from the European Collection of Cell Cultures (ECACC), UK. Hep G2 (human hepatocellular carcinoma cell line) and Caco-2 (human colorectal adenocarcinoma cell line) cells were maintained in DMEM medium (Sigma) supplemented with 10% FBS and 2 mM L-glutamine.44,45 PC-3 (human prostate carcinoma cell line), B16F10 (mouse melanoma cell line) and CT26 (mouse colon carcinoma cell line) cells were maintained in RPMI medium (Sigma) supplemented with 10% FBS.46–48 These cells (passage number < 30) were grown in the incubator (ThermoForma) at 37 °C with 5% CO2 and 95% relative humidity.The cytotoxicity of Au NPs was estimated using the MTT assay with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma).46 Hep G2 (20000 cells per well), Caco-2 (10000 cells per well), PC-3 (5000 cells per well), B16F10 (1000 cells per well) and CT26 (6000 cells per well) cells were seeded within 200 μL growth media in 96-well plates one day before transfection. Au NPs were incubated with cells for 24 h under the normal growth conditions. After incubation, the particle solution was replaced with 200 μL fresh growth medium, and 20 μL MTT stock (5 mg mL−1 in PBS) was added and incubated with cells for 4 h at 37 °C. The contents were removed and 100 μL DMSO was added to dissolve the purple formazan products. The data were obtained at 590 nm using a microplate reader. The concentration of Au NPs leading to 50% cell growth inhibition (IC50) was estimated from the plot of the percentage of viable cells versus the concentration of Au NPs for each treatment.
2.8. Preparation and characterisation of Au NP·siRNA complexes
A colloidal solution of Au NPs (100 μg mL−1) were added into an siRNA solution (siRNA, sense sequence 5′-UUC UCC GAA CGU GUC ACG U-3′, was obtained from QIAGEN, USA and prepared in RNase-free water following QIAGEN recommendations), at different mass ratios (MRs) of Au NPs to siRNA, followed by 1 h incubation with slightly shaking at RT. The ability of Au NPs to complex siRNA was analysed by gel retardation.49 Briefly, complexes of Au NPs and siRNA (containing 0.25 μg siRNA) at different mass rations (MRs) were loaded onto 1% (w/v) agarose gels in Tris/Borate/EDTA (TBE) buffer containing SafeView (NBS Biologicals, UK). Electrophoresis was performed at 120 V for 30 min and the resulting gels were photographed under UV. In addition, particle sizes and zeta potentials were measured with a Malvern Nano-ZS (Malvern Instruments, UK) at 25 °C using the default non-invasive back scattering (NIBS) technique with a detection angle of 173°.50 Deionised water (0.2 μm membrane-filtered, applied for particle size and zeta potential evaluation) were added to the complexes (Au·siRNA complexes at different MRs) and made up to 1 mL, before measurement. The concentration of siRNA was fixed at 1 μg mL−1.2.9. Fluorescence activated cell sorting
PC-3 cells (50000 per well) were seeded in 24-well plates and incubated for 24 h under normal growth conditions. Cells were then transfected by 20 nM fluorescein–siRNA complexed with GR11 (MR20) and incubated for 24 h in normal growth medium. Fluorescein-siRNA alone was used as the negative control, and fluorescein–siRNA complexed with INTERFRin™ (Polyplus) (prepared following the manufacturer's recommendation) was used as the positive control. On the following day, cells were first treated with CellScrub™ (Genlatins) to remove complexes associated with cell surfaces (uninternalised complexes) according to manufacturer's instructions. The medium was then removed, and cells were washed twice with PBS and trypsinised. Cells were subsequently centrifuged (1000 rpm for 5 min) and were re-suspended in 1000 μL cold PBS in polystyrene round-bottom tubes (Becton Dickinson). Ten thousands cells were measured for each sample following the application of Becton Dickinson FACScalibur manual. Fluorescein-positive cells (%) were displayed by Dot Plot.
2.10. Statistics
One-way analysis of variance (ANOVA) was used to compare multiple groups followed by Bonferroni's post hoc test. Statistical significance was set at *p < 0.05.3. Results and discussion
3.1. Synthesis and characterisation of positively charged Au NPs
The aim of our study was to form nearly spherical positively charged Au nanoparticles in water, with a wide mean diameter range, avoiding the use of toxic surfactants that may direct the final shape/size and biocompatibility of the nanoparticles. Typical synthesis methods for the production of positively charged gold nanoparticles employ the cationic surfactant CTAB as a capping and stabilising agent. CTAB forms a bilayer structure around Au NPs, with the inner layer binding to the Au surface via its charged head groups and the adsorbed bilayer leads to a net positive charge on the surface of the nanoparticles.51 These methods use very high concentration of CTAB that can often affect the final size and shape of the nanoparticles formed; for instance, CTAB is the most widely used surfactant for the synthesis of anisotropic multiple shapes of Au NPs such as nanorods or nanoprisms.52 Previous studies have shown that the presence of CTAB during seed-mediated growth methods leads to the formation of Au nanorods with aspect ratios ranging from 1.5 to 4.5,53 while the addition of a co-surfactant such as benzyldimethylhexadecylammonium chloride (BDAC) to CTAB yields the formation of a binary surfactant mixture that affects the aspect ratios of the Au nanorods, varying from 4.6 to 10.54 However, Au–CTAB nanoparticles are known to be highly toxic to different cell lines, due to the presence of excess surfactant.31,32,55 Our hypothesis in this study was that by producing colloidal solutions free of surfactants such as CTAB, low toxic Au NPs with a desired morphology can be obtained.21Ultra small Au–L-cysteine methyl ester seeds, with a mean diameter around 2 nm, were obtained by reducing AuCl3 with ice cold NaBH4 in the presence of L-cysteine methyl ester hydrochloride as a stabilising agent (Fig. S1†). The Au–L-cysteine methyl ester seeds obtained were further used to produce samples GR1 and GR2, by the surface catalysed reduction of AuCl3 with hydroxylamine hydrochloride, yielding an increased population of the seeds with mean diameters between 4–5 nm. As the cysteine solution contains hydrochloride, AuCl3 was chosen as a precursor in this study instead of tetrachloroauric acid HAuCl4 in order to avoid increasing the acidity of the aqueous solution, which would affect the synthesis process and the final particle size and aggregation state. Data in Table 1 and Fig. S1† obtained from TEM images show that nanoparticles generated in samples GR1 and GR2 were well below 10 nm (4 ± 1.5 nm and 5 ± 2 nm respectively) with a relatively high large size-polydispersity (PDI > 0.45); most likely due to the excess of NaBH4 used in the seed solutions causing the formation of some new Au(0) nuclei. The size polydispersity and diverse shape formation was reduced during synthesis, by growing the nanoparticles in a stepwise fashion.56–60 Therefore, for the rest of the gold colloidal solutions GR3–GR12 a successive growth method was used and GR2 seeds 5 ± 2 nm were ripened to ∼7 ± 2.5 nm (GR3), the latter was ripened to form GR4 nanoparticle solutions (diameters 9 ± 4 nm) that were ripened to form GR5 nanoparticle solutions (diameters 16 ± 2 nm) (Table 1). GR6 and GR7 nanoparticle solutions (diameters 46 ± 4 nm and 56 ± 4 nm respectively) were prepared from GR5 solutions by varying the concentration of the nanoparticle seeds. GR6 nanoparticle solutions (diameters 46 ± 4 nm) were ripened to 80 ± 6 nm (GR8 nanoparticle samples). GR9 and GR10 nanoparticle samples (diameters 104 ± 7 nm and 118 ± 9 nm respectively) were prepared from GR7 solutions by varying the concentration of the nanoparticle seeds, and finally GR11 and GR12 nanoparticle samples (diameters 145 ± 18 nm and 195 ± 20 nm respectively) were prepared from GR10 solutions by varying the concentration of the nanoparticle seeds. The successive growth method used in this study allowed us to obtain nearly spherical and size-monodisperse Au NPs–cysteine samples (GR5-GR12) as confirmed by SEM (Fig. 2) and DLS (0.07 < PDI < 0.2) analysis (Table 1).
| Sample name | λ max (nm) | Δλ (nm) | A max | Diameter (nm) EM | Standard dev. | Hydrodynamic diameter (nm) DLS | PDI | Zeta potential (mV) | Zeta deviation (mV) |
|---|---|---|---|---|---|---|---|---|---|
| Seeds | — | — | — | 1.9 | 0.4 | — | — | 38.3 | 14.6 |
| GR1 | 519 | 70 | 0.78 | 4 | 1.5 | 4.2 | 0.46 | 34.8 | 11.5 |
| GR2 | 521 | 86 | 0.94 | 5.5 | 2 | 5.1 | 0.55 | 35.1 | 14 |
| GR3 | 521 | 89 | 0.96 | 7 | 3 | 24 | 0.6 | 34.8 | 17.1 |
| GR4 | 522 | 91 | 1.12 | 9 | 4.5 | 7 | 0.5 | 33.2 | 18 |
| GR5 | 524 | 89 | 1.15 | 16 | 2 | 19 | 0.2 | 34 | 14.3 |
| GR6 | 629 | 87 | 1.09 | 46 | 4 | 47 | 0.14 | 34.9 | 18.1 |
| GR7 | 533 | 93 | 0.98 | 56 | 4 | 55 | 0.11 | 37.6 | 16.4 |
| GR8 | 554 | 126 | 1.16 | 80 | 6 | 86 | 0.07 | 45.4 | 13.5 |
| GR9 | 572 | 160 | 0.94 | 104 | 7 | 104 | 0.07 | 44.3 | 11.8 |
| GR10 | 576 | 171 | 0.8 | 118 | 9 | 117 | 0.07 | 49 | 12.3 |
| GR11 | 607 | 251 | 0.56 | 145 | 18 | 139 | 0.10 | 41.2 | 10.8 |
| GR12 | 2 peaks | >380 | ∼0.2 | 195 | 20 | 195 | 0.15 | 41.6 | 11.1 |
3.2. UV-visible absorption spectroscopy, EM and DLS
Table 1 summarises the UV-visible absorption spectroscopy (Fig. 1) and either TEM or SEM Fig. S1† and 2 data obtained from the Au NPs. The UV-visible spectra of the seeds showed that the nanoparticles had no clear plasmon band, indicating that they were ultra-small in size, as the plasmon band disappears when the size of Au clusters approaches 2 nm (Fig. 1(a)).61 TEM data also verified that the mean diameter of the seeds was around 1.9 nm (σ = 0.4 nm) (Fig. S1†). As the size of the nanoparticles was increased from 4 ± 1.5 nm (GR2) to ∼80 ± 6 nm (GR8) a slight increase in the absorbance from ∼0.78 to ∼1.15 (GR8) was observed, accompanied by a red shift in the plasmon band from 519 to 554 nm.40,62 However, when the size of the nanoparticles increased above 80 nm in diameter a decrease in the plasmon absorbance was noticed, i.e. from ∼1.15 for (GR8) to ∼0.56 (GR11), with a plasmon band red shift more prominent from 554 nm (GR8) to 607 nm (GR11) when the Au NP diameter increased from 80 ± 6 nm (GR8) to 145 ± 18 nm (GR11). The observed red shift was also accompanied by a further broadening of the absorbance band. The band width (λ0) was found to increase by about 24 nm from 70 to 91 ± 3 nm when the mean nanoparticle diameter increased from 4 nm (GR1) to 56 nm (GR7), and by approximately 147 nm from 94 to 251 nm when the mean nanoparticle diameter increased from 56 nm (GR7) to 145 nm (GR11). Finally, the GR12 nanoparticle solutions (diameters ∼ 195 nm) displayed poorly defined two peaks with a broad absorption between 520–900 nm (Fig. 1(b))40,62 The broadening of the absorbance band and peak shift became more pronounced when the size of the nanoparticles increased above 50 nm; this can be explained by the fact that higher oscillation modes (quadrupole, octopole absorption and scattering) may also affect the nanoparticle extinction cross sections with increasing size.62,63 The electron microscopy images in Fig. 2 and S1† show that the nanoparticle diameter distribution widths and circularity were not strongly affected by increasing the diameter (no nanorods or other particular shapes were detected) and therefore comparable diameter distribution widths and nanoparticle circularity to the initial spherical seeds were observed.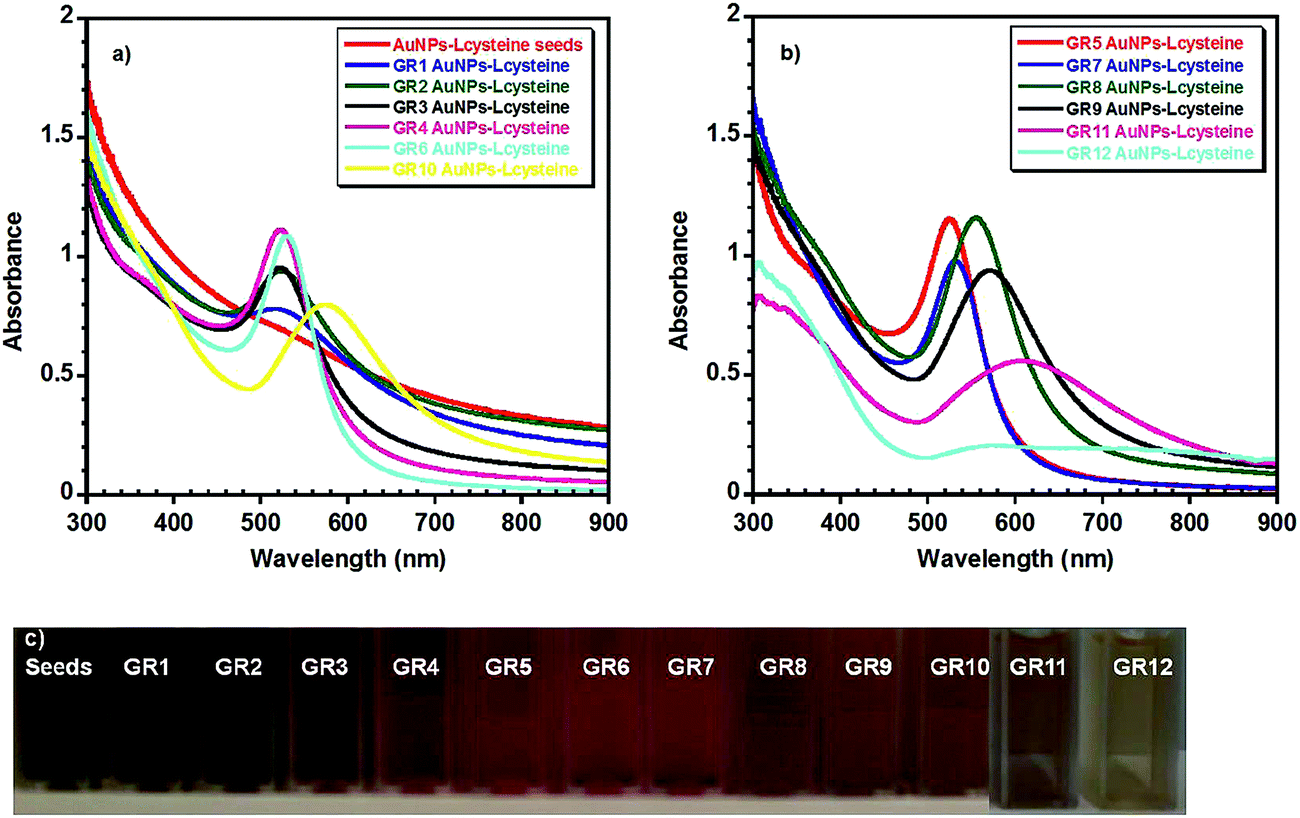 | ||
| Fig. 1 (a) and (b) UV-visible spectra of different Au NPs. The spectra were split for better visualisation. Samples in (b) were used in the cytotoxicity assays. (c) Optical photographs of the corresponding colloidal solutions. | ||
 | ||
| Fig. 2 SEM of Au NPs with mean diameters of (GR5) ∼ 16 nm, (GR7) ∼ 56 nm, (GR8) ∼ 80 nm, (GR9) ∼ 104 nm, (GR11) ∼ 145 nm and (GR12) ∼ 195 nm. | ||
3.3. Dynamic light scattering
DLS, combined with electron microscopy can provide a full physical characterisation of colloidal dispersions of Au NPs for biomedical applications. The zeta potential measurements can also be measured by DLS. Fig. 3(a) shows the size distribution, by intensity, of samples GR5, GR7, GR8, GR9, GR11 and GR12 and Fig. 3(b) highlights the zeta potentials (mV) of all of the Au NPs–cysteine solutions synthesised in this study. For Au NP samples with diameters below 10 nm (GR1–GR4), size distributions were difficult to measure by DLS; samples GR1–GR4 were slightly size-polydisperse with a polydispersity index (PDI) around 0.53 ± 0.06. However, larger nanoparticle samples GR5 (16 nm) to GR12 (195 nm) were nearly size-monodisperse with a PDI around 0.13 ± 0.06, and a measured hydrodynamic diameter (Dh) very close to the diameters obtained from EM analysis (see Table 1). The zeta potential of all of the nanoparticle samples prepared in this study showed a positive value around +39 ± 5 (Fig. 3(b) and S2†) indicating that the nanoparticles were successfully coated with a positively charged capping layer of L-cysteine methyl ester hydrochloride (0.5 mmol L−1), which provided colloidal stability. | ||
| Fig. 3 (a) Size distribution by intensity for the Au NP–L-cysteine methyl ester hydrochloride (GR5, GR7 to GR12, these were used in the cytotoxicity assays). (b) Zeta Potential for Au NPs (G1 to G12). | ||
3.4. Cytotoxicity
A comparison of the cytotoxicity of Au nanoparticle solutions of GR5, GR7, GR8, GR9, GR11 and GR12 with Au NPs–CTAB samples was performed using a range of human and murine cells (Fig. 4 and Table S1†). The cytotoxicity of Au NPs was studied using an MTT assay. In the PC-3 cell line, GR5, GR7, GR8, GR9, GR11 and G12 samples displayed IC50 values of 8.2, 7.8, 8.2, 7.4, 8.5 and 8.0 μg mL−1 respectively. In contrast, Au NP–CTAB samples (20 and 60 nm) caused a higher level of cytotoxicity with IC50 values of 2 and 3.5 μg mL−1 respectively (Table S1†). In addition, similar data were observed in Hep G2, Caco-2, B16F10 and CT26 cells (Table S1†). Goodman et al. have previously reported that positively charged CTAB-stabilised Au NPs may cause higher cytotoxicity compared to their anionic carboxylate stabilised counterparts. Here we have demonstrated that Au NP–CTAB samples (20 and 60 nm) caused higher cell death compared to non-CTAB Au–L-cysteine nanoparticles (Fig. 4 and Table S1†). | ||
| Fig. 4 Viability (MTT) of PC-3 cells treated with Au NPs (mean ± SD, n = 3). | ||
Cationic Au nanoparticles in general have been reported to cause high toxicity,64 and demonstrate in vivo instability due to aggregation followed by non-specific uptake by the mononuclear phagocytic system (MPS, also known as the reticuloendothelial system (RES)).65 One of the best characterised methods to improve the biocompatibility and in vivo stability of cationic Au NPs is the addition of polyethylene-glycol (PEG, a highly soluble, non-toxic and non-immunogenic polymer, approved for use by the Food and Drug Administration (FDA)).
3.5. Formation of Au NP·siRNA complexes
Recently, Au NPs have been functionalised by grafting cationic polymers and biomolecules onto the surface of gold in order to bind negatively charged nucleic acid via electrostatic interaction achieving a complex.16,17,66 In addition, thiol (SH)-functionalised nucleic acids have also been used to attach nucleic acid onto Au NPs thus forming a complex due to the high affinity of thiol to gold to produce AuNP–thiolates (Au–S).67–70 In this study siRNA was chosen to examine the capacity of Au NPs to deliver nucleic acids. The formation of Au NP·siRNA complexes through the interaction between the positively charged amino groups on the surface of the Au NPs and negatively charged siRNA was examined by gel retardation (Fig. 5). As can be seen from Fig. 5 the complete complexation of siRNA with GR11 nanoparticle samples took place from a mass ratio (MR) of 20 onwards. The complexation of siRNA using Au NPs with smaller diameters (<100 nm) was not as efficient as with larger NPs (>100 nm) (Fig. S3†), suggesting that siRNA did not easily complex around small particles because of the stiff rod-like structure of siRNA.71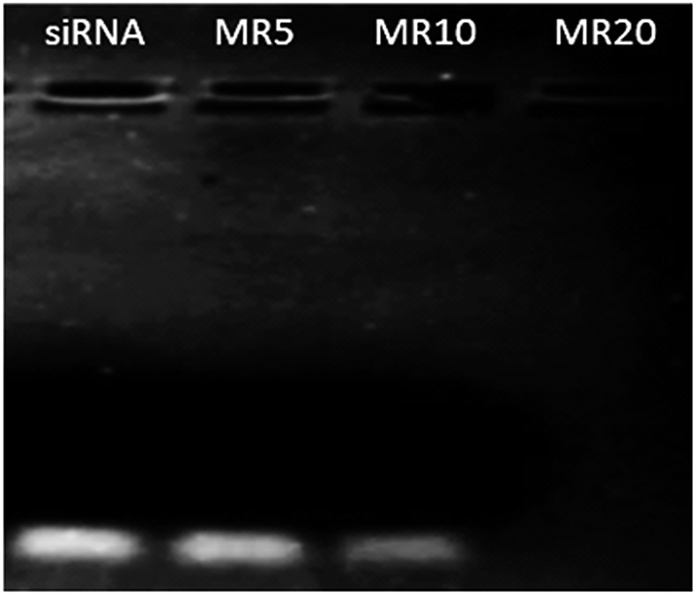 | ||
| Fig. 5 Complexation of siRNA (0.25 μg) with GR11 samples at different mass ratios (MRs of Au NPs to siRNA, MR = 5, 10 and 20). 0.25 μg of siRNA was used as negative control. | ||
The GR11·siRNA complex at MR20 demonstrated a cationic surface (∼+25 mV), in comparison with negatively charged counterparts at MR5 and 10 (∼−22 and −11 mV, respectively), confirming that siRNA was efficiently complexed with GR11 at MR20 onwards (Fig. 5). In addition, the mean diameters of GR11·siRNA complexes at MR20 in DI water were in the diameter range of 155 ± 15 nm; whereas, a significant increase in size (>1000 nm) occurred for GR11·siRNA complexes at MR5 and MR10. These data suggest that the positively charged formulation effectively prevented aggregation in comparison with the negatively charged counterparts, indicating that cationic materials may remain stable in solution.72 However, previous studies have shown that aggregation of positive complexes is time dependent.73 In addition to improving cell viability, PEGylation has been reported to stabilise complexes against salt-, protein- and complement-induced inactivation by steric effects as well as by shielding surface charges.39,74,75 Future studies will produce PEGylated Au–L-cysteine nanoparticles and will examine the physicochemical, in vitro and in vivo profiles of these vectors for siRNA delivery.
3.6. Internalisation of Au NP·siRNA complexes
Fluorescein–siRNA was used to study the internalisation of Au NP·siRNA complexes in PC-3 cells. The fluorescein–siRNA formulated with GR11 (MR20) significantly increased cellular uptake compared to fluorescein–siRNA alone; in addition, GR11 achieved similar cellular uptake (up to 20%) compared to INTERFERin™ (up to 25%) (Fig. 6; the representative Dot Plots showing fluorescein-positive cells (%) are shown in Fig. S4†), suggesting that the Au NPs were able to deliver nucleic acids into the cells. In addition, further bioconjugation via the ammonium group and hydroxyl protected (o-methyl ester) functional group on the surface of these Au NPs can be used to improve intracellular trafficking of nucleic acids into the cytoplasm, in the case of antisense oligodeoxynucleotide (ODN), siRNA and microRNA (miRNA), or the nucleus, in the case of plasmid DNA and short hairpin RNA (shRNA), with the overall aim of regulating gene expression.76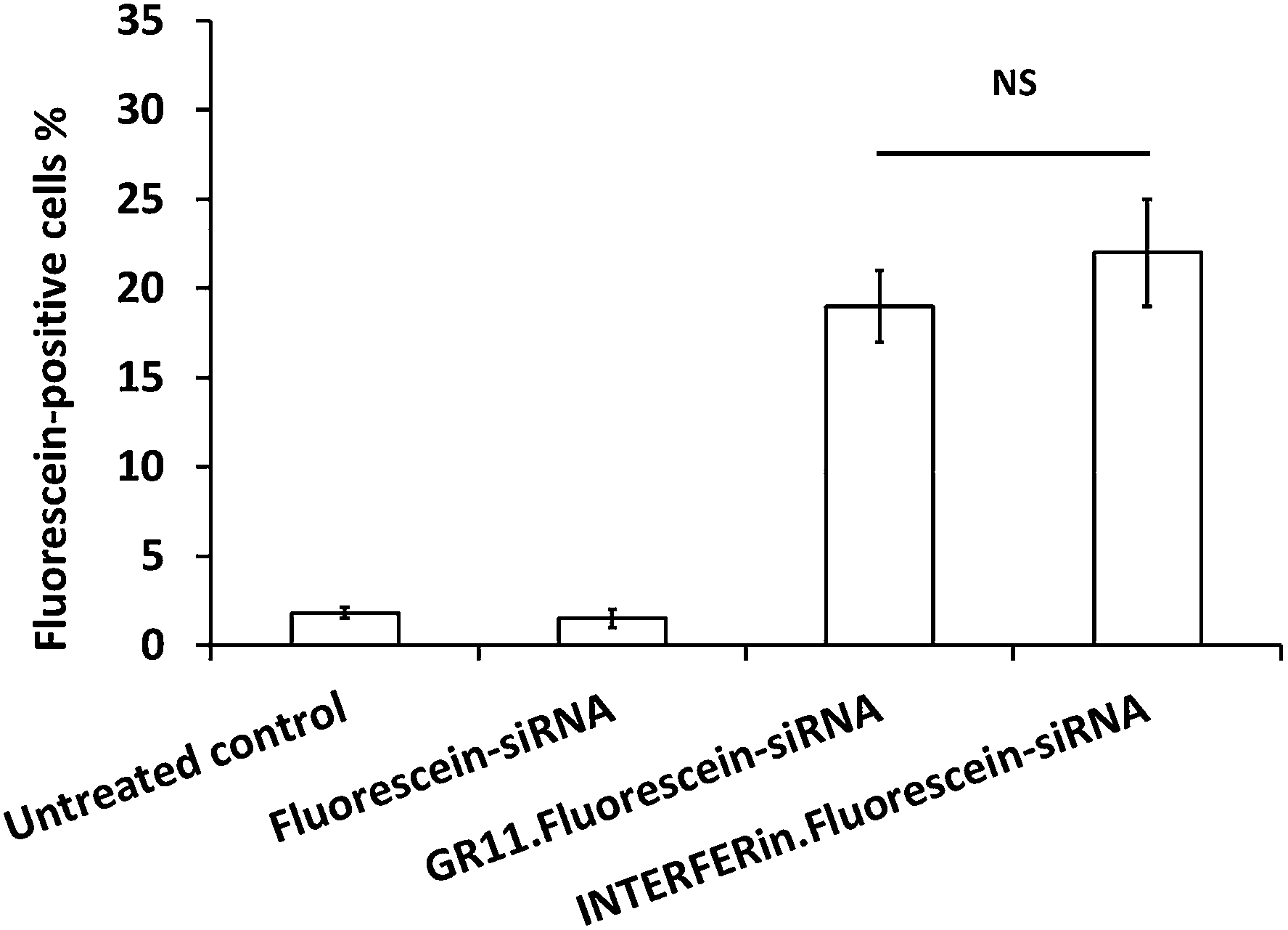 | ||
| Fig. 6 Fluorescein-positive cells (%) with 20 nM fluorescein–siRNA, either uncomplexed and complexed with GR11 (MR20) and INTERFERin™, analysed by Dot Plot in PC-3 cells (NS = no significance) (mean ± SD, n = 3). | ||
4. Conclusions
Positively charged, surfactant-free and nearly spherical Au NPs have been synthesised in water with a wide range of diameters (2 to 200 nm) and narrow diameter distributions 10–15% (with the exception for the sizes below 10 nm), using a controlled seed mediated growth approach. These Au NPs showed size-dependent optical properties making them very suitable for photonic and bioimaging applications. The presence of L-cysteine methyl ester hydrochloride (HSCH2CH(NH2)COOCH3·HCl) on their surface with a mercapto group (–SH) anchoring the cysteine to the nanoparticles and an ammonium group (–NH3+) formed by the presence of an amine group in slightly acidic media confer positive charges to the resulting Au NP as confirmed from the zeta potential measurements (ζ-potential +33 to +49 mV). In addition, cytotoxicity studies performed in a range of human and murine cell lines demonstrated that the surfactant-free positively charged Au NPs synthesised in this study were less cytotoxic than Au–CTAB nanoparticles. Furthermore, Au NPs with diameters over 100 nm formed effective complexes with nucleic acids. In comparison with commercially available transfection reagent, these complexes demonstrated similar capacity to deliver nucleic acids, in terms of cellular uptake of fluorescein siRNA. Moreover, the presence of the ammonium group and hydroxyl protected (o-methyl ester) functional group on the surface of these Au NPs indicates the potential of Au NPs–L-cysteine methyl ester for further chemical bioconjugation to facilitate active targeting, receptor mediated delivery and improved intracellular trafficking in vitro and in vivo. Studies to evaluate the physicochemical properties, and the in vitro and in vivo performance of PEGylated and functionalised Au–L-cysteine nanoparticles as non-viral vectors for nucleic acid delivery are now in progress.Acknowledgements
We acknowledge financial from the Irish Research Council, for a Government of Ireland Postdoctoral Fellowship to Jianfeng Guo (GOIPD/2013/150). Microscopy analysis was undertaken at the Electron Microscopy and Analysis Facility (EMAF) at the Tyndall National Institute, Cork, Ireland.References
- L. C. Cheng, X. Jiang, J. Wang, C. Chen and R. S. Liu, Nanoscale, 2013, 5, 3547–3569 RSC.
- Y. Luo, M. Hossain, C. M. Wang, Y. Qiao, J. C. An, L. Y. Ma and M. Su, Nanoscale, 2013, 5, 687–694 RSC.
- S. H. Wen, K. G. Li, H. D. Cai, Q. Chen, M. W. Shen, Y. P. Huang, C. Peng, W. X. Hou, M. F. Zhu, G. X. Zhang and X. Y. Shi, Biomaterials, 2013, 34, 1570–1580 CrossRef CAS PubMed.
- G. Barhate, M. Gautam, S. Gairola, S. Jadhav and V. Pokharkar, Int. J. Pharm., 2013, 441, 636–642 CrossRef CAS PubMed.
- L. J. Jing, X. L. Liang, Z. J. Deng, S. S. Feng, X. D. Li, M. M. Huang, C. H. Li and Z. F. Dai, Biomaterials, 2014, 35, 5814–5821 CrossRef CAS PubMed.
- L. Q. Shao, J. J. Diao, Z. P. Tang, S. Liu, S. C. Shen, J. K. Liu, X. F. Rui, D. P. Yu and Q. Zhao, Nanoscale, 2014, 6, 4089–4095 RSC.
- A. Latorre, C. Posch, Y. Garcimartin, A. Celli, M. Sanlorenzo, I. Vujic, J. Ma, M. Zekhtser, K. Rappersberger, S. Ortiz-Urda and A. Somoza, Nanoscale, 2014, 6, 7436–7442 RSC.
- K. Mallick and M. J. Witcomb, Gold Nanoparticles: Properties, Characterization and Fabrication, 2010, 225–243 CAS.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- A. Kumar, X. Zhang and X. J. Liang, Biotechnol. Adv., 2013, 31, 593–606 CrossRef CAS PubMed.
- H. X. Cai and P. Yao, Nanoscale, 2013, 5, 2892–2900 RSC.
- J. B. Song, Z. Fang, C. X. Wang, J. J. Zhou, B. Duan, L. Pu and H. W. Duan, Nanoscale, 2013, 5, 5816–5824 RSC.
- A. Latorre, C. Posch, Y. Garcimartin, S. Ortiz-Urda and A. Somoza, Chem. Commun., 2014, 50, 3018–3020 RSC.
- D. W. Kim, J. H. Kim, M. Park, J. H. Yeom, H. Go, S. Kim, M. S. Han, K. Lee and J. Bae, Biomaterials, 2011, 32, 2593–2604 CrossRef CAS PubMed.
- D. R. Bhumkar, H. M. Joshi, M. Sastry and V. B. Pokharkar, Pharm. Res., 2007, 24, 1415–1426 CrossRef CAS PubMed.
- W. H. Kong, K. H. Bae, S. D. Jo, J. S. Kim and T. G. Park, Pharm. Res., 2012, 29, 362–374 CrossRef CAS PubMed.
- S. H. Lee, K. H. Bae, S. H. Kim, K. R. Lee and T. G. Park, Int. J. Pharm., 2008, 364, 94–101 CrossRef CAS PubMed.
- S. K. Lee, M. S. Han, S. Asokan and C. H. Tung, Small, 2011, 7, 364–370 CrossRef CAS PubMed.
- S. Y. Moon, T. Kusunose and T. Sekino, Mater. Lett., 2009, 63, 2038–2040 CrossRef CAS.
- R. Narayanan, R. J. Lipert and M. D. Porter, Anal. Chem., 2008, 80, 2265–2271 CrossRef CAS PubMed.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383–1396 RSC.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, small, 2009, 5, 646–664 CrossRef CAS PubMed.
- Y. Huang, W. Wang, H. Liang and H. Xu, Cryst. Growth Des., 2009, 9, 858–862 CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS PubMed.
- A. M. Alkilany, P. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- M. Tarantola, A. Pietuch, D. Schneider, J. Rother, E. Sunnick, C. Rosman, S. Pierrat, C. Sönnichsen, J. Wegener and A. Janshoff, Nanotoxicology, 2011, 5, 254–268 CrossRef CAS PubMed.
- T. Mironava, M. Hadjiargyrou, M. Simon and M. H. Rafailovich, Nanotoxicology, 2014, 8, 189–201 CrossRef CAS PubMed.
- C. S. Yah, Biomed. Res., 2013, 24, 400–413 CAS.
- C. J. Johnson, E. Dujardin, S. A. Davis, C. J. Murphy and S. Mann, J. Mater. Chem., 2002, 12, 1765–1770 RSC.
- R. Chen, J. Wu, H. Li, G. Cheng, Z. Lu and C. M. Che, Rare Met., 2010, 29, 180–186 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613–11618 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- T. Zhao, X. Shen, L. Li, Z. Guan, N. Gao, P. Yuan, S. Q. Yao, Q. H. Xu and G. Q. Xu, Nanoscale, 2012, 4, 7712–7719 RSC.
- M. S. Dobrick, K. V. Sarathy and M. Jansen, J. Am. Chem. Soc., 2005, 127, 12816–12817 CrossRef PubMed.
- J. Patel, L. Němcová, P. Maguire, W. G. Graham and D. Mariotti, Nanotechnology, 2013, 24, 245604 CrossRef CAS PubMed.
- W. J. Song, J.-Z. Du, T.-M. Sun, P. Z. Zhang and J. Wang, Small, 2009, 6, 239–246 CrossRef PubMed.
- K. Esumi, N. Takei and T. Yoshimura, Colloids Surf., B, 2003, 32, 117–123 CrossRef CAS.
- A. Elbakry, E. C. Wurster, A. Zaky, R. Liebl, E. Schindler, P. B. Kreisel, T. Blunk, R. Rachel, A. Goepferich and M. Breunig, Small, 2012, 8, 3847–3856 CrossRef CAS PubMed.
- K. Rahme, M. T. Nolan, T. doody, G. P. McGlacken, M. A. Morris, C. O'Driscoll and a. J. D. Holmes, RSC Adv., 2013, 3, 21016–21024 RSC.
- K. Rahme, L. Chen, R. G. Hobbs, M. A. Morris, C. O'Driscoll and a. J. D. Holmes, RSC Adv., 2013, 3, 6085–6094 RSC.
- K. R. Brown and M. J. Natan, Langmuir, 1998, 14, 726–728 CrossRef CAS.
- K. R. Brown, L. A. Lyon, A. P. Fox, B. D. Reiss and M. J. Natan, Chem. Mater., 2000, 12, 314–323 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- S. J. R. Meex, U. Andreo, J. D. Sparks and E. A. Fisher, J. Lipid Res., 2011, 52, 152–158 CrossRef CAS PubMed.
- M. J. O'Neill, J. F. Guo, C. Byrne, R. Darcy and C. M. O'Driscoll, Int. J. Pharm., 2011, 413, 174–183 CrossRef PubMed.
- J. Guo, W. P. Cheng, J. Gu, C. Ding, X. Qu, Z. Yang and C. O'Driscoll, Eur. J. Pharm. Sci., 2012, 45, 521–532 CrossRef CAS PubMed.
- X. F. He, J. Wang, J. Dou, F. L. Yu, K. Cai, X. L. Li, H. Y. Zhang and N. Gu, Exp. Ther. Med., 2011, 2, 911–916 Search PubMed.
- F. Y. Huang, Y. N. Li, W. L. Mei, H. F. Dai, P. Zhou and G. H. Tan, Asian Pac. J. Trop. Med., 2012, 5, 169–174 CrossRef CAS PubMed.
- J. Guo, A. M. O'Mahony, W. P. Cheng and C. M. O'Driscoll, Int. J. Pharm., 2013, 447, 150–157 CrossRef CAS PubMed.
- J. Guo, J. R. Ogier, S. Desgranges, R. Darcy and C. O'Driscoll, Biomaterials, 2012, 33, 7775–7784 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- G. Oza, S. Pandey, R. Shah, M. Vishwanathan, R. Kesarkar, M. Sharon and M. Sharon, Adv. Appl. Sci. Res., 2012, 3, 1027–1038 CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- C. K. Wang, J. E. Wang, D. J. Liu and Z. X. Wang, Talanta, 2010, 80, 1626–1631 CrossRef CAS PubMed.
- L. Cao, T. Zhu and Z. Liu, J. Colloid Interface Sci., 2006, 293, 69–76 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- S. Hong and X. Li, J. Nanomater., 2013, 790323 Search PubMed.
- K. Mallick, Z. L. Wang and T. Pal, J. Photochem. Photobiol., A, 2001, 140, 75–80 CrossRef CAS.
- C. Ziegler and A. Eychmuller, J. Phys. Chem. C, 2011, 115, 4502–4506 CAS.
- Y. G. Kim, S. K. Oh and R. M. Crooks, Chem. Mater., 2004, 16, 167–172 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- N. G. Khlebtsov and L. A. Dykman, J. Quant. Spectrosc. Radiat. Transfer, 2010, 111, 1–35 CrossRef CAS.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- J. Guo, L. Bourre, D. M. Soden, G. C. O'Sullivan and C. O'Drisocll, Biotechnol. Adv., 2011, 29, 402–417 CrossRef CAS PubMed.
- R. Ghosh, L. C. Singh, J. M. Shohet and P. H. Gunaratne, Biomaterials, 2013, 34, 807–816 CrossRef CAS PubMed.
- J. Conde, F. Tian, Y. Hernández, C. Bao, D. Cui, K. P. Janssen, M. R. Ibarra, P. V. Baptista, T. Stoeger and J. M. de la Fuente, Biomaterials, 2013, 34, 7744–7753 CrossRef CAS PubMed.
- P. Resnier, T. Montier, V. Mathieu, J.-P. Benoit and C. Passirani, Biomaterials, 2013, 34, 6429–6443 CrossRef CAS PubMed.
- S. Borse, S. Joshi and A. Khan, RSC Adv., 2015, 5, 13402–13410 RSC.
- D. Zheng, D. A. Giljohann, D. L. Chen, M. D. Massich, X.-Q. Wang, H. Iordanov, C. A. Mirkin and A. S. Paller, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11975–11980 CrossRef CAS PubMed.
- A. Elbakry, A. Zaky, R. Liebkl, R. Rachel, A. Goepferich and M. Breunig, Nano Lett., 2009, 9, 2059–2064 CrossRef CAS PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- E. Lai and J. H. v. Zanten, Biophys. J., 2001, 80, 864–873 CrossRef CAS PubMed.
- J. V. Jokerst, T. Lobovkina, R. N. Zare and S. S. Gambhir, Nanomedicine, 2011, 6, 715–728 CrossRef CAS PubMed.
- K. Rahme, F. Gauffre, J. D. Marty, B. Payre and C. Mingotaud, J. Phys. Chem. C, 2007, 111, 7273–7279 CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16294c |
| This journal is © The Royal Society of Chemistry 2015 |