
Mechanical properties of structurally-defined magnetoactive polymer (co)networks†
Fotios Mpekrisa,
Mariliz Achilleosb,
Eugenia Vasilec,
Eugeniu Vasilec,
Theodora Krasia-Christoforoub and
Triantafyllos Stylianopoulos*a
aCancer Biophysics Laboratory, Department of Mechanical and Manufacturing Engineering, University of Cyprus, P.O. Box 20537, Nicosia, 1678, Cyprus. E-mail: tstylian@ucy.ac.cy; Fax: +357 2289 5081; Tel: +357 2289 2238
bPolymers Laboratory, Department of Mechanical and Manufacturing Engineering, University of Cyprus, P.O. Box 20537, Nicosia, 1678, Cyprus
cDepartment of Oxide Materials and Nanomaterials, Faculty of Applied Chemistry and Material Science, University Politehnica of Bucharest, No. 1-7 Gh. Polizu Street, 011061 Bucharest, Romania
First published on 12th February 2015
Abstract
The mechanical properties of structurally-defined magnetoactive polymer (co)networks synthesized from well-defined poly(2-dimethylamino)ethyl methacrylate (poly(DMAEMA)) homopolymers and diblock copolymers of poly(DMAEMA) with the hydrophobic n-butyl methacrylate (BuMA) were measured in compression. Magnetic nanoparticle composition varied from 0 to 30% wt and caused a 6-fold increase in the Young's modulus of the homopolymer networks (2.91 vs. 18.62 kPa) and a 12-fold increase in the modulus of diblock copolymer networks (0.76 vs. 9.1 kPa), with homopolymers being stiffer. Mathematical modeling revealed an exponential constitutive equation to predict accurately the mechanical response of the polymers. Furthermore, experiments were performed for the poroelastic behavior of the materials and their hydraulic conductivity was found to be independent of magnetic loading and network structure. In conclusion, the incorporation of magnetic nanoparticles strengthened the (co)network structure, while the synthetic approach employed for the DMAEMA-b-BuMA formation retained the linear, non-crosslinked architecture of BuMA, resulting in less stiff structures.
Introduction
Polymer networks are 3-dimensional (3-D) structures, consisting of polymer chains interconnected via chemical or physical crosslinks.1 Their crosslinked architecture prevents their dissolution in organic or aqueous solvents enabling only their swelling.2,3 Polymer networks prepared by non-controlled chemical crosslinking processes such as conventional free radical polymerization, often suffer from morphological inhomogeneities and poor mechanical and swelling properties.4For overcoming the above-mentioned limitations and for allowing the structure-to-property correlation, new synthetic approaches have been proposed towards the generation of structurally-defined polymer networks. These include “quasi-living” carbocationic polymerization,5,6 anionic polymerization,7,8 group transfer polymerization (GTP)9,10 and controlled radical polymerization processes such as atom transfer radical polymerization (ATRP)11–14 and reversible addition fragmentation chain transfer (RAFT) polymerization.15,16 The aforementioned polymerization methods lead to well-defined polymer chains with precisely controlled molecular weights (MW), chemical composition and narrow molecular weight distribution (MWD) that are interconnected with chemical crosslinks forming a 3-D network architecture.
Very recently we have reported on the synthesis of polymer (co)networks, with pre-defined and controlled architectures involving the crosslinking of well-defined poly(2-dimethylamino)ethyl methacrylate (poly(DMAEMA)) homopolymers and polyDMAEMA-containing block copolymers prepared by RAFT, using 1,2-bis-(2-iodoethoxy)-ethane (BIEE) as a crosslinker. Besides the synthesis of functional polymer (co)networks with various chemical structures and compositions, this new synthetic approach enables the generation of polymer-based nanocomposite (co)networks with structurally defined characteristics, via the encapsulation of inorganic nanoparticles (NPs) such as magnetic iron oxide (Fe3O4) NPs during the crosslinking step.17
Although the synthesis arm for the generation of structurally-defined polymer networks has been well-developed, the characterization of these materials in regards to their mechanical properties and the interconnection of the mechanical response to the network structural and compositional characteristics have not been extensively studied. Only a few literature reports appear so far dealing with this aspect,18 whereas in most cases the focus is given on the swelling behavior of these materials5,15,16,19,20 and on microphase separation phenomena occurring within the network structure, with the latter applying in the case of structurally-defined amphiphilic conetworks.2 In one example where the authors studied the mechanical properties of ethylene glycol dimethacrylate (EGDMA)-crosslinked amphiphilic and ionizable model co-networks prepared by GTP, the co-network uniaxial compression modulus of the water-swollen co-networks was found to be affected by the solution pH (due to the presence of the ionizable DMAEMA units) whereas it increased linearly with the increase in the hydrophobic n-butyl methacrylate (BuMA) content.21 In another work, high-strength polyethylene glycol (PEG)-containing hydrogels were prepared by combining two symmetrical tetrahedron-like macromonomers with four PEGylated arms of equal length, possessing mutually reactive end groups.22 The authors demonstrated that the fabrication of a homogeneous gel structure dictated by the controlled length of the tetrahedral PEG arms within the two macromonomers, results in a high mechanical strength that can be further enhanced upon tuning the PEG arm length.
Herein the mechanical properties of structurally-defined BIEE-crosslinked DMAEMA homopolymers and DMAEMA-b-BuMA diblock copolymer (co)networks and of their Fe3O4 nanocomposites are investigated for the first time. Besides the fact that the mechanical properties of this new type of structurally-defined polymer (co)networks have not been previously studied, to the best of our knowledge this is the first report on the investigation of the mechanical properties of magnetoresponsive polymer (co)networks with structural-defined characteristics. To this end, we synthesized DMAEMA homopolymer and DMAEMA-b-BuMA diblock copolymer networks enriched with Fe3O4 nanoparticles with compositions spanning from 0% to 30% wt. The mechanical properties of these (co)networks were tested under compressive loading conditions and two different types of experiments were performed: stress–strain and stress relaxation to study the elastic and poroelastic behavior of the materials, respectively. The experimental results were further analyzed with the use of mathematical modeling. An exponential hyperelastic constitutive equation was used to predict the elastic response of all network structures and fitting this equation to the experimental data, the corresponding material constants were derived for each network type. Finally, by employing a biphasic, poroelastic model, the hydraulic conductivities of the materials were calculated.
Results and discussion
Morphological characterization of the nanocomposite (co)networks
The scope of this work was the study of the effect of the chemical composition and of the magnetic loading on the mechanical behavior of structurally-defined polymer (co)networks. Pre-formed OA·Fe3O4 NPs, were loaded in two different polymer structures: the poly(2-(dimethylamino)ethyl methacrylate) (DMAEMA) homopolymer network and the poly(2-(dimethylamino)ethyl methacrylate)-block-poly(n-butyl methacrylate) (DMAEMA-b-BuMA) block copolymer network. Fig. 1 provides the chemical structures of the two monomers (DMAEMA, BuMA) and the crosslinking agent (BIEE) used for the synthesis of the above-mentioned systems and shows schematically the crosslinking process taking place in the absence and presence of OA·Fe3O4. | ||
| Fig. 1 Chemical structures of the two monomers and the crosslinking agent (BIEE) employed in the synthesis of the (co)networks and schematic illustration of the DMAEMA homopolymer and the DMAEMA-b-BuMA diblock copolymer crosslinking process, taking place in the absence and presence of OA·Fe3O4 NPs. | ||
High-resolution transmission electron microscopy (HR-TEM) analysis was employed for obtaining information on the geometrical characteristics i.e. size and shape and the crystalline structure adopted by the magnetic nanoparticles embedded within the polymeric matrix, as well as the morphology of the nanocomposites. From the TEM images appearing in Fig. 2, the magnetic nanoparticles embedded within the networks appear spherical in shape with average diameters of ∼7 nm (nanoparticle size distribution histograms are provided in ESI, Fig. 1†). Moreover, a distinct dispersion of the NP within the polymer matrix resulting in high homogeneity was observed in all cases. Selected area electron diffraction (SAED) for crystalline structure evaluation was also performed. SAED images show a face centered cubic crystalline structure analogous magnetite (Fe3O4), in accordance with ICDD file no. 04-008-8146. Energy dispersive X-ray spectroscopy (spectra are provided in the ESI, Fig. 2†) reveals the presence of the iron, iodine (due the presence of the iodine-containing BIEE crosslinker), carbon and oxygen elements in the samples. The presence of Cu is attributed to the Cu grid employed in TEM investigations.
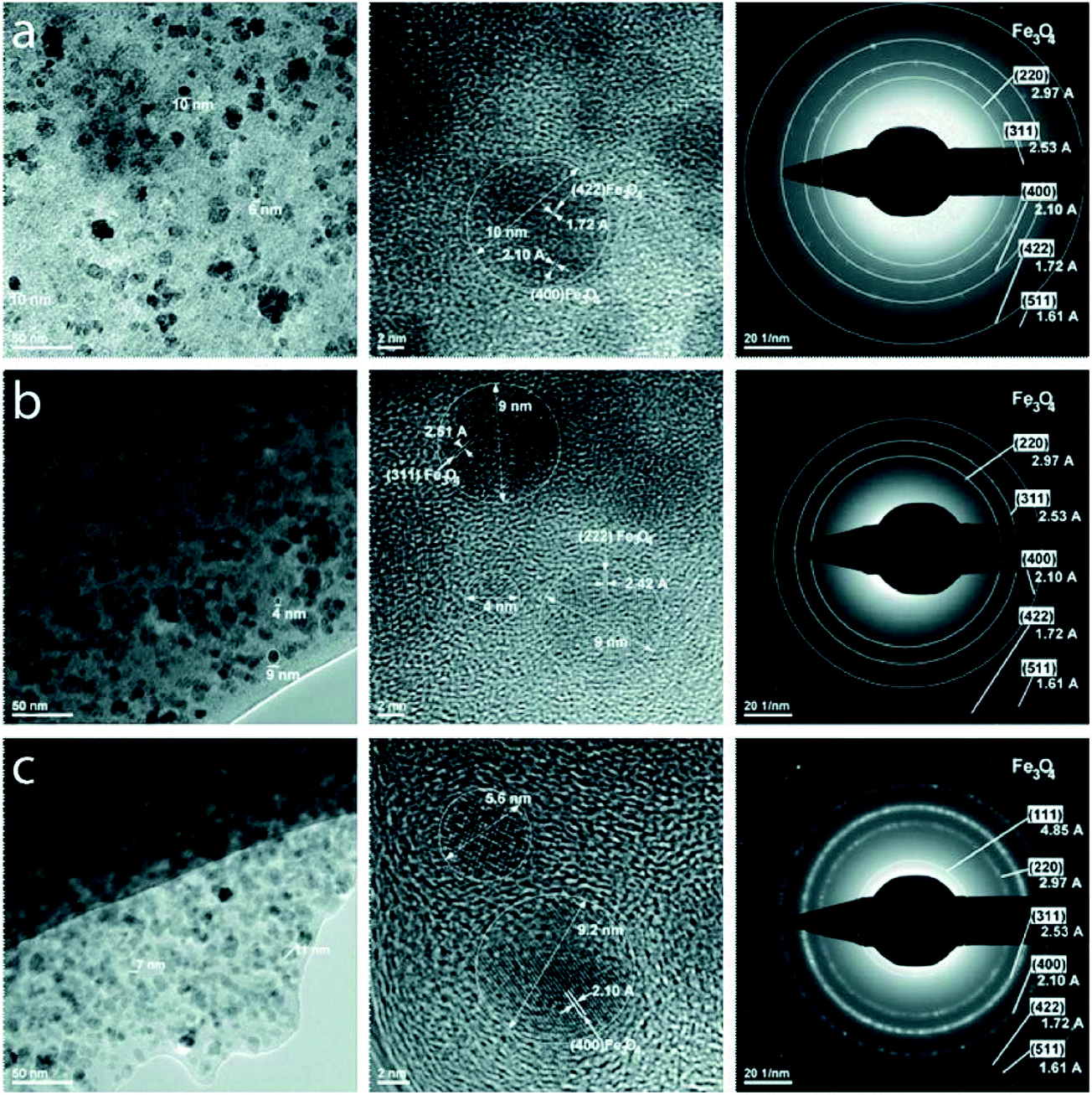 | ||
| Fig. 2 HR-TEM images of the nanocomposite networks. Left and middle columns: (a) DMAEMA (10%) (b) DMAEMA/BuMA (10%) and (c) DMAEMA/BuMA (30%) at different magnification levels. Scale bars: 50 nm (left column), 2 nm (middle column). Right column: selected area electron diffraction (SAED) images. | ||
Based on thermal analysis data reported in our previous publication17 the thermal stability of the DMAEMA homopolymer networks and the DMAEMA-containing block copolymer conetworks exhibits a slight improvement in the presence of OA·Fe3O4 nanoparticles in comparison to their pristine polymer analogues, suggesting the existence of polymer–Fe3O4 nanoparticle interactions23 with the nanoparticles acting as heat barriers, thus affecting favorably the thermal stability of the networks.
Mechanical behavior of the DMAEMA and DMAEMA-b-BuMA (co)networks in the absence of OA·Fe3O4
As suggested in Fig. 1, the synthetic methodology employed, results in the formation of structurally-defined DMAEMA-b-BuMA conetworks, in which the BuMA polymer chains retain their linear architecture after the crosslinking process. A comparison of the stress–strain experimental curves (Fig. 3) as well as the measured mechanical properties (Fig. 4) of the DMAEMA homopolymer and the DMAEMA-b-BuMA diblock copolymer networks in the absence of magnetic nanoparticles shows that the homopolymer networks are stiffer than the copolymers, presumably due to the inability of BuMA to resist compressive loads. Indeed, BuMA chains are not crosslinked to each other or to DMAEMA chains and thus, they can deform freely. When subjected to compressive loads these chains will buckle and will not contribute to the mechanical response of the network. Interestingly, in a different study DMAEMA-b-BuMA conetworks were prepared by free-radical solution polymerization leading to random network structures with enhanced mechanical properties.24 Therefore, the polymerization conditions and the network structure (structurally-defined or random) determines in large part the mechanical properties of the networks. | ||
| Fig. 3 Stress–strain experimental curves. The figure depicts the stress–strain response of the (A) DMAEMA and (B) DMAEMA-b-BuMA networks. Addition of magnetic nanoparticles increases the applied stress on the specimens making the polymers stiffer. Numbers in parentheses denote the percent of magnetic loading. | ||
 | ||
| Fig. 4 Young's modulus derived from the experimental stress–strain curves. The Young's modulus is greater when magnetic nanoparticles are added in the homopolymer and diblock copolymer networks. | ||
Modulation of the (co)network mechanical properties with magnetic loading
The effect of the addition of magnetic nanoparticles on the homopolymer networks is presented in Fig. 3A, while Fig. 3B presents the effect of magnetic loading on the mechanical response of the block copolymers. In both cases, it is clear that loading with OA·Fe3O4 NPs stiffens the polymer networks and the increase in the stiffness is higher for higher magnetic content. The same conclusion is reached from Fig. 4, which shows the Young's modulus of the different polymer networks, calculated as the slope of the linear part of the stress–strain curves. The nanoparticles form composites with the crosslinked DMAEMA block (Fig. 1), which adds to the strength of the network.18 Furthermore, the block copolymers are softer than the homopolymer networks for the same magnetic loading, in line with the observed mechanical response of the corresponding pristine polymer networks as discussed above. This is expected since during crosslinking the magnetite nanoparticles are entrapped within the DMAEMA domains whereas the BuMA chains retain their linear architecture thus providing further flexibility to the network. Statistical analysis of our data comparing all possible pairs within the same polymeric group or between the two polymeric groups for the same magnetic loading reveals that there is a statistically significant difference for all cases (ESI, Table 1†).Simulation of the network mechanical properties
As shown in Fig. 3, the mechanical response of the homopolymer and block copolymer networks is highly nonlinear. Based on this observation, an exponential constitutive equation (eqn (1)) was employed to simulate the stress–strain experiments and fit the constitutive equation to the experimental data. Representative fits for all six cases tested are presented in Fig. 5. From the fits it is clear that the exponential expression can accurately predict the experimental behavior of the networks. The values of the mechanical properties derived by fitting the model to the experimental results are summarized in Table 1. | ||
| Fig. 5 Representative fits of the exponential constitutive equation (eqn (1)) to the data for the (A) DMAEMA and (B) DMAEMA-b-BuMA networks. | ||
| Polymer type | A1 (kPa) | A2 (kPa) | C1 | χ2 |
|---|---|---|---|---|
| DMAEMA | 0.01 ± 0.01 | 66.3 ± 7.9 | 26.4 ± 5.7 | 0.22 ± 0.04 |
| DMAEMA + 10% OA·Fe3O4 | 0.12 ± 0.05 | 98.8 ± 27.3 | 33.0 ± 10.3 | 0.38 ± 0.03 |
| DMAEMA + 30% OA·Fe3O4 | 7.47 ± 2.6 | 228.3 ± 61.1 | 14.4 ± 3.1 | 0.51 ± 0.1 |
| DMAEMA-b-BuMA | 0.001 ± 0.0002 | 12.7 ± 2.3 | 30.6 ± 1.1 | 0.19 ± 0.03 |
| DMAEMA-b-BuMA + 10% OA·Fe3O4 | 0.51 ± 0.2 | 56.6 ± 6.1 | 21.6 ± 2.9 | 0.45 ± 0.08 |
| DMAEMA-b-BuMA + 30% OA·Fe3O4 | 1.42 ± 1.1 | 46.5 ± 12.3 | 44.3 ± 11.3 | 0.43 ± 0.04 |
Investigation of the poroelastic behavior of the networks
To investigate the poroelastic response of the two polymer groups as well as the effect of the magnetic loading on it, stress relaxation experiments were performed. Representative stress relaxation results are shown in Fig. 6. The results are typical of viscoelastic/poroelastic materials. When the strain is applied with a fast rate 5% min−1 the total stress reaches a maximum, which is the sum of the stresses of the solid and fluid phase. Subsequently, the specimen is held constant for 10 min, the fluid pressure vanishes and the stress reaches an equilibrium point, which corresponds to the contribution of the solid stress alone. Because the co-polymer network is soft, the solid stress vanishes completely or even gets negative values at the first two cycles (i.e., up to 10% strain) and it becomes resistant to the compression at higher strains, during the last two cycles of the experimental procedure. Fitting this stress relaxation behavior to the poroelastic model the hydraulic conductivity of the materials was calculated (Fig. 7). Fig. 7 suggests that magnetic loading, even though affected the mechanical behavior of the polymers significantly, has no effect on the hydraulic conductivity.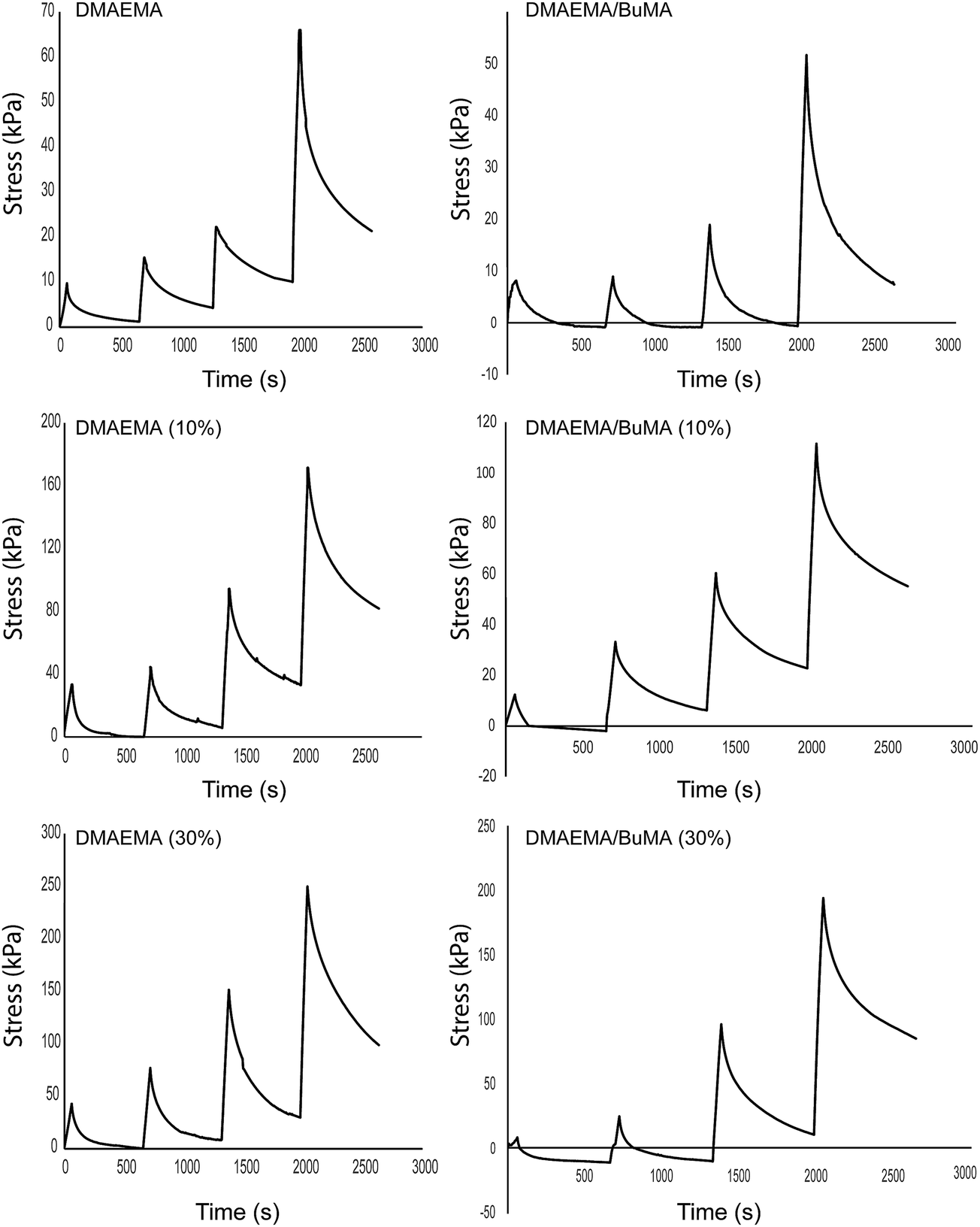 | ||
| Fig. 6 Representative results of the stress relaxation experiments. The specimens were subjected to four cycles of 5% strain. Numbers in parentheses denote the percent of magnetic loading. | ||
 | ||
| Fig. 7 Hydraulic conductivity of the networks. Fitting a poroelastic model to the stress relaxation experimental data the hydraulic conductivity was determined. | ||
Conclusions
In this study, the mechanical properties of DMAEMA homopolymer and DMAEMA-b-BuMA diblock copolymer networks enriched with Fe3O4 nanoparticles with compositions spanning from 0% to 30% wt were investigated. Homopolymer networks were stiffer than the diblock copolymer networks presumably owing to the fact that in the latter, the BuMA polymer chains retained their linear architecture after crosslinking, thus enabling them to deform freely upon compression. Importantly, a strong correlation between magnetic nanoparticle loading and the elastic properties was observed for both homopolymer and diblock copolymer structures. There was no correlation, however, between nanoparticle loading and the hydraulic conductivity of the networks. Mathematical analysis of the experimental data suggested that an exponential constitutive equation was appropriate to predict the elastic response of all network structures.Experimental
Materials and methods
Benzene (Fluka, >99%) was stored over CaH2 (Merck, 95%) and was distilled under reduced pressure prior to the polymerization reactions. 2-(Dimethylamino)ethyl methacrylate (DMAEMA, Merck, 99%) and n-butyl methacrylate (n-BuMA, Merck, 99%) were passed through a basic alumina column (Activated, basic, Brockmann I, ∼150 mesh, pore size 58 Å) stored over CaH2 and distilled under reduced pressure immediately prior to the polymerization reactions. 2,2-Azobis(isobutyronitrile) (AIBN) (Sigma-Aldrich, 95%) was recrystallized from ethanol and dried under vacuum at room temperature. 2-Cyano-2-propyldithiobenzoate (CPDTB, Sigma-Aldrich, 97%), 1,2-bis-(2-iodoethoxy)ethane (BIEE, Sigma-Aldrich, 96%), n-hexane (Scharlau, 96%) and tetrahydrofuran (THF) (Scharlau, 99.9%) were used as received.Characterization
The MWs and MWDs of the polymers were determined by size exclusion chromatography (SEC) using equipment supplied by Polymer Standards Service (PSS). All measurements were carried out at room temperature using Styragel HR 3 and Styragel HR 4 columns. The mobile phase was THF, delivered at a flow rate of 1 ml min−1 using a Waters 515 isocratic pump. The refractive index was measured with a Waters 2414 refractive index detector supplied by PSS. The instrumentation was calibrated using poly(methyl methacrylate) (PMMA) standards with narrow MWD (MWs of 739![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 446000, 270000, 126000, 65000, 31000, 14400, 4200, 1580, 670, 450, and 102 (methyl isobutyrate) g mol−1) supplied by PSS. 1H NMR spectra were recorded in CDCl3 with tetramethylsilane (TMS) used as an internal standard using an Avance Brucker 300 MHz spectrometer equipped with an Ultrashield magnet.
000, 446000, 270000, 126000, 65000, 31000, 14400, 4200, 1580, 670, 450, and 102 (methyl isobutyrate) g mol−1) supplied by PSS. 1H NMR spectra were recorded in CDCl3 with tetramethylsilane (TMS) used as an internal standard using an Avance Brucker 300 MHz spectrometer equipped with an Ultrashield magnet.
The geometrical characteristics (size and shape) and the crystalline structure adopted by the magnetic nanoparticles as well as the morphology of the nanocomposite networks were investigated by high-resolution transmission electron microscopy (HR-TEM) using a TECNAI F30 G2 S-TWIN microscope operated at 300 kV with Energy Dispersive X-ray Analysis (EDX) facility. For TEM investigations, a small amount of sample was milled in an agate mortar and diluted in water. After sonicating for 30 min, the solution was deposited on a TEM copper grid covered with a thin amorphous carbon film with holes. After drying the TEM grids were placed on a sample holder and the samples could be studied by transmission electron microscopy.
Synthetic procedures
937 g mol−1, Mw: 12067 g mol−1, polydispersity index (PDI): 1.10) (ESI, Fig. 3†).1H NMR: δH (300 MHz; CDCl3; Me4Si): 0.8–1.1 (3H, m, –CH3), 1.5–2.00 (2H, m, –CH2), 2.34 (3H, s, –NCH3), 2.56 (2H, s, –NCH2), and 4.05 (2H, s, –OCH2) (ESI, Fig. 4†).
937 g mol−1, 5 g, 0.45 mmol of the macroCTA) and n-BuMA (1.3 g, 9.1 mmol) were added into a round bottom flask (50 ml) equipped with a PTFE stirring bar. AIBN (0.044 g, 0.27 mmol) was dissolved in benzene (6.8 ml) and was added into the reaction flask with the aid of a syringe. The reaction mixture was stirred rapidly at room temperature, degassed by three freeze–evacuate–thaw cycles and heated at 65 °C for 20 hours. The polymerization was terminated by cooling the reaction mixture down to room temperature. The produced polyDMAEMA-b-polyBuMA was retrieved by precipitation in n-hexane (50 ml) and dried at room temperature under vacuum (yield: 72%, Mn: 15230 g mol−1, Mw: 17916 g mol−1, PDI: 1.18) (ESI, Fig. 3†).1H NMR: δH (300 MHz; CDCl3; Me4Si): 0.85–1.10 (3H, m, –CH3), 1.30–2.10 (2H, m, –CH2), 2.33 (3H, s, –NCH3), 2.64 (2H, s, –NCH2), 3.93 (2H, s, –OCH2 BuMA), and 4.09 (2H, s, –OCH2 DMAEMA) (ESI, Fig. 4†).
937 g mol−1, 0.20 g, 0.0183 mmol of macroCTA, 1.27 mmol of DMAEMA units) was dissolved in THF (2.5 ml). To the polymer solution, BIEE (0.117 ml, 0.237 g, 0.64 mmol, 0.5 mol per DMAEMA unit) was added using a micropipette. The mixture was stirred rapidly for a few seconds using a vortex mixer and then left in the vial (sealed so as to prevent THF from evaporating) without stirring at room temperature. Crosslinking was observed after 5 days.230 g mol−1, 0.2 g, 0.00132 mmol of macroCTA, 0.906 mmol of DMAEMA units) was dissolved in THF (2.5 ml). To the polymer solution, BIEE (0.083 ml, 0.168 g, 0.453 mmol, 0.5 mol per DMAEMA unit) was added using a micropipette. The mixture was stirred rapidly for a few seconds using a vortex mixer and then left in a sealed glass vial without stirring at room temperature. Crosslinking was observed after 9 days.937 g mol−1, 0.20 g, 0.0183 mmol of macroCTA, 1.27 mmol of DMAEMA units). After complete solubilization of the polymer in the THF solution, BIEE (0.117 ml, 0.237 g, 0.64 mmol, 0.5 mol per DMAEMA unit) was added using a micropipette. The mixture was stirred rapidly for a few seconds using a vortex mixer and then left without stirring at room temperature. Crosslinking was observed after 6 days.230 g mol−1, 0.2 g, 0.00132 mmol of macroCTA, 0.906 mmol of DMAEMA units). When the polymer was dissolved, BIEE (0.083 ml, 0.168 g, 0.453 mmol, 0.5 mol per DMAEMA unit) was added using a micropipette. The mixture was stirred rapidly for a few seconds using a vortex mixer and then left without stirring at room temperature. Crosslinking was observed after 7 days.Determination of sol-fraction
After crosslinking, the networks were removed from the vial and were left to equilibrate in THF (100 ml) for 1 week to remove the sol fraction (extractables). Afterwards, the solvent was recovered by filtration and evaporated. The recovered extractables were dried under vacuum at room temperature for one day. The sol-fraction was determined gravimetrically and was calculated from the ratio of the dried mass of the extractables to the theoretical mass of all components in the network (i.e., polymer plus crosslinker plus OA·Fe3O4 where applicable). Extractable percentages varied between 6–13%.Mechanical testing experiments
Unconfined compression experiments were performed using a high precision mechanical testing system (Instron 5944, Norwood, MA, USA). The specimens (pre-swelled in tetrahydrofuran) were cut in an orthogonal shape with dimensions 4 × 4 × 2 mm (length × width × thickness). According to the unconfined compression experiment the specimen was placed between two parallel platens, while two different compression protocols were employed. For the first protocol, a strain-controlled, stress–strain experiment was performed to test the elastic response of the materials. The specimens were compressed to 25% strain with a strain rate of 0.05 mm min−1, the lowest strain rate that the system can apply, in order to avoid any transient, poroelastic effects. The stress was calculated as the force measured on the load cell divided by the initial area of the specimen (i.e., 1st Piola–Kirchhoff stress) and the strain was calculated as the displacement Δl divided by the initial length of the specimen. The Young's modulus was calculated from the slope of the linear part of the stress–strain curves. Five specimens from each material and condition were tested (n = 5). The second experimental procedure involved stress relaxation experiments to study the poroelastic behavior of the materials. According to the stress relaxation protocol, the specimens underwent four cycles of testing for each of which a 5% strain was applied within a period of a minute followed by a ten minutes hold. As in the stress–strain experiment, n = 5 for these experiments as well.Mathematical analysis of experimental data
Mathematical modeling was employed to further analyze the mechanical properties of the polymer (co)networks. Specifically, a finite elements model was employed to simulate the stress–strain experiments, and subsequently the finite elements model was extended to account for the fluid phase using a biphasic, poroelastic theory to simulate the stress relaxation experiments (details in ESI†).For the elastic model, an exponential, hyperelastic constitutive equation was employed following our previous research with a strain energy density function of the form:26,27
| W = A1(eC1(−3+II1) − 1) + A2(−1 + J)2, | (1) |
 . A three-dimensional finite elements model of orthogonal geometry, same in size as that of the real specimens, was constructed. The model was compressed in one direction and was free to deform in the other two according to the unconfined compression experimental protocol. The material properties of the constitutive equation (i.e., A1, A2 and C1) were varied so that the sum of the squared errors,
. A three-dimensional finite elements model of orthogonal geometry, same in size as that of the real specimens, was constructed. The model was compressed in one direction and was free to deform in the other two according to the unconfined compression experimental protocol. The material properties of the constitutive equation (i.e., A1, A2 and C1) were varied so that the sum of the squared errors,  , reached a minimum. Pexp, Pmodel are the experimentally measured and predicted by the model 1st Piola–Kirchhoff stresses, respectively and n the number of experimental data.
, reached a minimum. Pexp, Pmodel are the experimentally measured and predicted by the model 1st Piola–Kirchhoff stresses, respectively and n the number of experimental data.
For the biphasic poroelastic model, eqn (1) was used for the solid phase and the fluid phase was assumed to be described by the hydrostatic pressure p, the hydraulic conductivity K and the fluid velocity, vf, through Darcy's law: vf = −K∇p (details in ESI, Fig. 3†). The total stress of the material is the sum of the stress of the solid phase minus the hydrostatic pressure of the fluid phase. A finite elements model was constructed to simulate the stress relaxation experiment. The constants A1, A2 and C1 of the solid phase that were found by fitting the elastic model to the stress–strain experiments were used, while the hydraulic conductivity, i.e., the easiness with which the fluid percolates through the pores of the polymer matrix, was varied so that the sum of the squared errors of the total experimental and modeled stresses to be minimized. Therefore fitting the model to the stress–strain data provided the elastic material properties of the polymers, while fitting of the stress-relaxation data provided the values of the hydraulic conductivity.
Statistical analysis
The data are presented as means with standard errors. Experimental groups were compared using an unpaired Student's t-test. Statistical significant difference was determined when p < 0.05.Acknowledgements
This work is part of the Project TEXNOΛOΓIA/YΛIKA/0311(BIE)/03 that is co-funded by the European Regional Development Fund and the Republic of Cyprus through the Cyprus Research Promotion Foundation. FM was supported by the University of Cyprus under the Programme “New Researchers”. The authors wish to thank Ladislau Vekas and Alina Moaca for providing the OA·Fe3O4 NPs and Maria Demetriou for the synthesis of the linear DMAEMA-b-BuMA diblock copolymer.References
- C. S. Patrickios, Special Issue: Polymer Networks: Synthesis, Properties, Theory and Applications, Macromol. Symp., 2010, 291–292, 1–11 CrossRef CAS.
- C. S. Patrickios and T. K. Georgiou, Curr. Opin. Colloid Interface Sci., 2003, 8, 76–85 CrossRef CAS.
- A. Noro, M. Hayashi and Y. Matsushita, Soft Matter, 2012, 8, 6416–6429 RSC.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120 CrossRef CAS.
- T. K. Georgiou, C. S. Patrickios, P. W. Groh and B. Ivan, Macromolecules, 2007, 40, 2335–2343 CrossRef CAS.
- K. Suvegh, A. Domjan, G. Vanko, B. Ivan and A. Vertes, Macromolecules, 1998, 31, 7770 CrossRef.
- H. P. Hentze, E. Kramer, B. Berton, S. Forster, M. Antonietti and M. Dreja, Macromolecules, 1999, 32, 5803 CrossRef CAS.
- L. Bromberg, M. Temchenko and T. A. Hatton, Langmuir, 2002, 18, 4944 CrossRef CAS.
- A. I. Triftaridou, S. C. Hadjiyannakou, M. Vamvakaki and C. S. Patrickios, Macromolecules, 2002, 35, 2506–2513 CrossRef CAS.
- A. I. Triftaridou, D. Kafouris, M. Vamvakaki, T. K. Georgiou, T. C. Krasia, E. Themistou, N. Hadjiantoniou and C. S. Patrickios, Polym. Bull., 2007, 58, 185–190 CrossRef CAS.
- F. Asgarzadeh, P. Ourdouillie, E. Beyou and P. Chaumont, Macromolecules, 1999, 32, 6696–7002 CrossRef.
- H. Gao, W. Li and K. Matyjaszewski, Macromolecules, 2008, 41, 2335–2340 CrossRef CAS.
- M. D. Rikkou, M. Kolokasi, K. Matyjaszewski and C. S. Patrickios, J. Polym. Sci., 2010, 48, 1878 CrossRef CAS.
- J. Xu, M. Qiu, B. Ma and C. He, ACS Appl. Mater. Interfaces, 2014, 6, 1528–1529 Search PubMed.
- T. C. Krasia and C. S. Patrickios, Macromolecules, 2006, 39, 2467–2473 CrossRef CAS.
- M. Achilleos, T. Krasia-Christoforou and C. S. Patrickios, Macromolecules, 2007, 40, 5575–5581 CrossRef CAS.
- M. Achilleos, M. Demetriou, O. Marinica, L. Vekas and T. Krasia-Christoforou, Polym. Chem., 2014, 5, 4365 RSC.
- W. Richteringa and B. R. Saunders, Soft Matter, 2014, 10, 3695 RSC.
- Z. Y. Lu and R. Hentschke, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 66, 041803 CrossRef.
- M. R. Simmons, E. F. Yamasaki and C. S. Patrickios, Macromolecules, 2000, 33, 3176–3179 CrossRef CAS.
- M. Vamvakaki and C. S. Patrickios, Soft Matter, 2008, 4, 268–276 RSC.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- E. Goiti, M. M. Salinas, G. Arias, D. Puglia, J. M. Kenny and C. Mijangos, Polym. Degrad. Stab., 2007, 92, 2198–2205 CrossRef CAS.
- A. Emileh, E. Vasheghani-Farahani and M. Imani, Eur. Polym. J., 2007, 43, 1986–1995 CrossRef CAS.
- D. Bica, Rom. Rep. Phys., 1995, 47, 265–272 CAS.
- C. Voutouri, F. Mpekris, P. Papageorgis, A. D. Odysseos and T. Stylianopoulos, PloS One, 2014, 9, e104717 Search PubMed.
- Y. C. Fung, Biomechanics: mechanical properties of living tissues, Springer-Verlag, New York, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16260a |
| This journal is © The Royal Society of Chemistry 2015 |