
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature synthesis of macroporous LiTi2(PO4)3/C with superior lithium storage properties
Hany
El-Shinawi
*ab and
Jürgen
Janek
a
aInstitute of Physical Chemistry, Justus-Liebig-University, Heinrich-Buff-Ring 58, 35392 Giessen, Germany. E-mail: H_elshinawi@mans.edu.eg; Fax: +49-641-99-34509; Tel: +49-641-99-34514
bChemistry Department, Faculty of Science, Mansoura University, Mansoura 35516, Egypt
First published on 23rd January 2015
Abstract
We report a “low-temperature” LiTi2(PO4)3 (LTP) phase (LT-LTP) with modified structural, microstructural and electrochemical properties. A macroporous LT-LTP phase is prepared by a simple sol–gel procedure followed by calcination at 550 °C. LT-LTP exhibits an unusual distribution of lithium ions within the NASICON-type structure and, consequently, a new two-step lithium-intercalation regime. The material, after compositing with carbon, shows excellent lithium storage properties with a retained capacity >100 mA h g−1 after 800 cycles at a 10 C charge–discharge rate. Macroporous LT-LTP/C composites exhibit a remarkably enhanced cycle performance compared to conventional LTP/C composites.
1. Introduction
LiTi2(PO4)3 (LTP) and LTP/C composites are currently being investigated as promising anode materials for aqueous lithium ion batteries1–6 and hybrid supercapacitors.7,8 The combination with carbon enhances the electronic conductivity of the material making it possible to utilize the advantageous properties of LTP electrodes, such as, excellent electrode potential in an aqueous electrolyte (∼2.5 V versus Li+/Li, −0.5 V versus NHE), flat voltage plateau, good specific capacity of 138 mA h g−1, low cost of the raw material, and environmental benignity. The electrochemical behavior of LTP/C and, particularly, good cycling stability in aqueous electrolytes, have encouraged researchers to develop several LTP/C composites/architectures with improved lithium storage properties.9–11 Different syntheses of LTP/C so far have adopted a relatively high calcination temperature (800–1000 °C) followed by a carbon-coating step.1–13 This produces a phase with a conventional NASICON-type structure where PO4 tetrahedra and TiO6 octahedra are corner-shared creating two types of crystal sites for lithium ions, commonly referred to as M1 and M2; the M1 sites are situated between pairs of TiO6 octahedra along the c-axis, while the M2 sites are located between infinite ribbons of [O3TiO3M1O3TiO3⬚O3TiO3M1]∞ (Fig. 1). Lithium in LiTi2(PO4)3 selectively occupies the M1 sites. Lithium intercalation in the material, however, is achieved by filling of the M2 cavities, which is associated with cooperative migration of Li+ ions from the M1 to the M2 sites, till the Li3Ti2(PO4)3 phase is eventually reached.14,15 This leads to a single (two-phase) lithium uptake step with a current/voltage plateau at ∼2.4 V versus Li+/Li. | ||
| Fig. 1 Part of the NASICON-type crystal structure showing the M1 and M2 crystal sites. | ||
Here we report “low-temperature” LTP phases with modified structural/electrochemical properties and excellent cycle performance. We have employed a modified sol–gel approach to synthesize macroporous LTP at calcination temperature as low as 550 °C. The material is composed of 3D interconnected nanoparticles (50–100 nm size) which provide high surface area, large surface-to-volume ratio, and favorable structural stability of the electrode material. Low-temperature LTP, moreover, exhibits a new two-step Li intercalation regime with redox peaks at ∼2.8 and 2.4 V. After compositing with carbon, the material exhibits a superior cycle performance compared to conventional LTP/C composites.
2. Materials and methods
A modified sol–gel procedure was employed to synthesize LiTi2(PO4)3. Lithium acetate (99.95%), titanium isopropoxide (≥97%) and lithium dihydrogen phosphate (≥98.0%) were purchased from Aldrich and used as received as starting materials. Lithium acetate was first dissolved in acetic acid and then mixed with EDTA (dissolved in NH3 solution) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio. Dilute NH3 solution was used to adjust the pH value of the solution to a value ∼5. The resulting solution was then carefully added to a stoichiometric amount of titanium isopropoxide. After addition of few drops of H2O2 solution (30 wt%), a stoichiometric amount of lithium dihydrogen phosphate was then added slowly with continuous stirring. The resulting clear solution was evaporated at ∼110 °C to produce a transparent gel, which was subsequently heated at 250 °C to form a black solid precursor. The solid precursor was ground and finally calcined in air at 550 °C for 24 h. The as-prepared LTP phase is abbreviated as “LT-LTP” to indicate that the material is synthesized at low calcination temperature. To composite LT-LTP with carbon (3–4 wt%), stoichiometric amount of the material was dispersed in a sucrose solution with a suitable concentration. The solvent was then evaporated slowly at 80 °C under constant stirring. The resulting solid precursor was then dried at 80 °C in vacuum, followed by calcination at 550 °C for 3 h in Ar atmosphere.
:3 molar ratio. Dilute NH3 solution was used to adjust the pH value of the solution to a value ∼5. The resulting solution was then carefully added to a stoichiometric amount of titanium isopropoxide. After addition of few drops of H2O2 solution (30 wt%), a stoichiometric amount of lithium dihydrogen phosphate was then added slowly with continuous stirring. The resulting clear solution was evaporated at ∼110 °C to produce a transparent gel, which was subsequently heated at 250 °C to form a black solid precursor. The solid precursor was ground and finally calcined in air at 550 °C for 24 h. The as-prepared LTP phase is abbreviated as “LT-LTP” to indicate that the material is synthesized at low calcination temperature. To composite LT-LTP with carbon (3–4 wt%), stoichiometric amount of the material was dispersed in a sucrose solution with a suitable concentration. The solvent was then evaporated slowly at 80 °C under constant stirring. The resulting solid precursor was then dried at 80 °C in vacuum, followed by calcination at 550 °C for 3 h in Ar atmosphere.
X-ray diffraction (XRD) data were collected with an X'Pert Pro PANalytical diffractometer in reflection mode, using CuKα radiation. Rietveld refinement based on XRD data were performed using the GSAS suite of programs.16 Scanning electron microscopy (SEM) was performed using a MERLIN machine from Zeiss. Nitrogen physisorption experiments for surface area measurement were carried out at 77 K using the Autosorb-1-MP automated gas adsorption station from Quantachrome. Thermogravimetric (TG) measurements were performed using a Rheometric Scientific STA 1500 thermal analyzer. Transmission electron microscopy (TEM) images were taken by a CM30 STEM from Philips. Electrodes of LTP-C/Super P/PVDF (80:10:10 wt%) were prepared using an aluminum strip as the current collector. Electrochemical tests were carried out in the voltage range 1.5–3.5 V using Swagelok cells with 1 M LiPF6 in EC/DMC (1:1) as electrolyte and lithium foil as counter and reference electrodes.
3. Results and discussion
The XRD pattern of as-prepared LTP (LT-LTP) is shown in Fig. 2. The pattern was readily indexed on a rhombohedral unit cell (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group), consistent with a NASICON-type structure. In addition to rhombohedral Rc reflections, a set of minor reflections were also observed in the XRD pattern of LT-LTP (Fig. 2). These reflections, however, could not be assigned to impurity or unreacted phases. A similar set of minor reflections were observed in the XRD patterns of iron substituted LTP (Li1+xTi2−xFex(PO4)3).17 In Li1+xTi2−xFex(PO4)3, titanium is partially substituted by iron and lithium, where lithium is believed to distribute over both the M1 and M2 sites of the NASICON-type structure. The minor reflections in Li1+xTi2−xFex(PO4)3 were attributed to an orthorhombic LTP phase that coexists with rhombohedral LTP and has an extension that is correlated with x (the amount of introduced Li/Fe).17,18 As described below, the electrochemical behavior of LT-LTP suggests that lithium in this material is distributed over both the M1 and M2 sites of the NASICON-type structure; a related distortion of the LTP lattice may therefore account for the unknown minor peaks in the XRD pattern of LT-LTP. Structural refinement based on XRD data of LT-LTP (Fig. 3) confirms a rhombohedral symmetry (Rc space group) of the primary phase with a = 8.5141(3) Å and c = 20.875(1) Å. It was not possible to locate Li+ in the structure because the refinements were considerably insensitive to the occupancy of Li+ sites; Li+ was therefore constrained to occupy the M1 site. The refined unit cell parameters (a = 8.5141(3) Å, c = 20.875(1) Å) are close to those typically observed in conventional LTP phases, however, a significant expansion of the c-parameter can be observed (e.g. in ref. 15, a = 8.5110(1) Å, c = 20.843(4) Å). Vacancies on the M1 sites, as evidenced by electrochemical analysis (see below), may account for the observed elongation of the unit cell along the c-axis (i.e. elongation of the TiO6/LiO6 ribbons).
c space group), consistent with a NASICON-type structure. In addition to rhombohedral Rc reflections, a set of minor reflections were also observed in the XRD pattern of LT-LTP (Fig. 2). These reflections, however, could not be assigned to impurity or unreacted phases. A similar set of minor reflections were observed in the XRD patterns of iron substituted LTP (Li1+xTi2−xFex(PO4)3).17 In Li1+xTi2−xFex(PO4)3, titanium is partially substituted by iron and lithium, where lithium is believed to distribute over both the M1 and M2 sites of the NASICON-type structure. The minor reflections in Li1+xTi2−xFex(PO4)3 were attributed to an orthorhombic LTP phase that coexists with rhombohedral LTP and has an extension that is correlated with x (the amount of introduced Li/Fe).17,18 As described below, the electrochemical behavior of LT-LTP suggests that lithium in this material is distributed over both the M1 and M2 sites of the NASICON-type structure; a related distortion of the LTP lattice may therefore account for the unknown minor peaks in the XRD pattern of LT-LTP. Structural refinement based on XRD data of LT-LTP (Fig. 3) confirms a rhombohedral symmetry (Rc space group) of the primary phase with a = 8.5141(3) Å and c = 20.875(1) Å. It was not possible to locate Li+ in the structure because the refinements were considerably insensitive to the occupancy of Li+ sites; Li+ was therefore constrained to occupy the M1 site. The refined unit cell parameters (a = 8.5141(3) Å, c = 20.875(1) Å) are close to those typically observed in conventional LTP phases, however, a significant expansion of the c-parameter can be observed (e.g. in ref. 15, a = 8.5110(1) Å, c = 20.843(4) Å). Vacancies on the M1 sites, as evidenced by electrochemical analysis (see below), may account for the observed elongation of the unit cell along the c-axis (i.e. elongation of the TiO6/LiO6 ribbons).
 | ||
| Fig. 2 XRD patterns of LT-LTP and LT-LTP/C. A set of unknown minor reflections are labeled with asterisk symbol (*). | ||
 | ||
| Fig. 3 Observed (+), calculated and difference profiles (solid lines) for XRD data collected from LT-LTP (λ = 1.5406 and 1.5444 Å); wRp = 0.0403, Rp = 0.0304, χ2 = 1.903. | ||
The microstructure of LT-LTP was investigated by SEM and TEM (Fig. 4). The study reveals that LT-LTP has a macroporous structure composed of 3D interconnected nanoparticles (50–100 nm particle size) (Fig. 4a–c). We attribute the macroporous structure of LT-LTP materials to the employed synthesis procedure and low calcination temperature (550 °C) that effectively suppresses the particle growth. The material showed a specific surface area of 67.3 m2 g−1, according to the standard Brunauer–Emmett–Teller (BET) method, and a specific pore volume of 0.93 cm3 g−1, consistent with a macroporous material.
 | ||
| Fig. 4 SEM and TEM images of LT-LTP (a–c), and the TG data collected from LT-LTP/C in air at a heating rate of 5 °C min−1 (d). | ||
Fig. 2 shows that the XRD pattern of LT-LTP/C composite is similar to that of LT-LTP. Thermogravimetric (TG) analysis indicates that the carbon content in LT-LTP/C is ∼3.5 wt%. TG analysis was conducted in air up to 700 °C and a weight loss of 3.6 wt% was observed in the temperature range of 300–550 °C (Fig. 4d). The observed weight loss agrees well with the weight ratio of LTP/sucrose (10:1) used in the synthesis procedure. A TEM study of LT-LTP/C reveals that a nanocomposite of LT-LTP and carbon is formed (Fig. 5b). Fig. 5 shows TEM images of LT-LTP and LT-LTP/C. LT-LTP/C clearly comprises nonuniform (interconnected) LTP nanoparticles (∼100 nm particle size) surrounded by nanostructured carbon (∼10 nm particle size). Hence, compositing LT-LTP with carbon yields an architecture in which the pores in macroporous LT-LTP host clusters of nanostructured carbon. This will effectively enhance the electronic conductivity of the material and optimize the electrochemical activity of LiTi2(PO4)3.
 | ||
| Fig. 5 TEM images of LT-LTP (a) and LT-LTP/C (b). | ||
The electrochemical characterization of LT-LTP and LT-LTP/C was performed using a conventional organic electrolyte (LiPF6 in EC/DMC). Fig. 6a and b shows slow sweep cyclic voltammograms (CVs) of LT-LTP and LT-LTP/C, respectively. More defined oxidation–reduction peaks were observed in case of LT-LTP/C due to the improved electronic conductivity of the material. However, both CVs clearly show that, in addition to the oxidation–reduction effects conventionally observed at ∼2.4 V, a significant redox effect is observed at ∼2.8 V. This behavior is commonly observed for NASICON-type Mn0.5Ti2(PO4)3 and related phases,19,20 rather than for conventional LiTi2(PO4)3 phases. Since Li+ ions occupy the M1 sites in conventional LiTi2(PO4)3, the material exchanges lithium in one-step (at ∼2.4 V) due to insertion of lithium in the vacant M2 sites and subsequent cooperative migration of Li+ ions from M1 to M2 sites.14,15 NASICON-type Mn0.5Ti2(PO4)3, however, shows a different lithium-intercalation behavior. In this compound, half of the M1 sites are occupied by Mn2+ ions which lead to a lithium-uptake step at ∼2.8 V, corresponding to insertion of 0.5 Li+ (per unit formula) into vacant M1 sites, followed by a second step at ∼2.4 V due to insertion of further lithium into the M2 sites.19 The observed electrochemical behavior of LT-LTP hence suggests that some of the M1 sites are empty due to a partial disorder of lithium over the M1 and M2 sites. Similar to Mn0.5Ti2(PO4)3, a reduction peak at 2.75 V in LT-LTP will correspond to filling of the vacant M1 sites by incoming Li+, while the second reduction peak at 2.35 V will correspond to insertion of excess lithium into the M2 sites. The corresponding oxidation peaks are observed at 2.85 and 2.60 V, respectively. A very small unknown oxidation peak is reproducibly observed at ∼2.3 V. A typical discharge–charge profile of LT-LTP/C is shown in Fig. 6c, and is clearly consistent with cyclic voltammetry data. The specific capacity due to the high-voltage reduction step (at ∼2.8 V) is approximately 17% of the total specific capacity. Since the total discharge capacity of LTP corresponds to insertion of 2 Li per unit formula, the discharge at 2.8 V hence corresponds to insertion of ∼0.34 Li per unit formula. These results suggest that about one third of the M1 sites were vacant in LT-LTP. The study, indeed, emphasizes the fact that the electrochemical analysis is a powerful tool to study the structural properties of this type of materials. It is worth mentioning that the occupancy of Li+ sites in the presence of heavier atoms (e.g. Ti) is a problematic structural feature that is barely detectable by XRD.
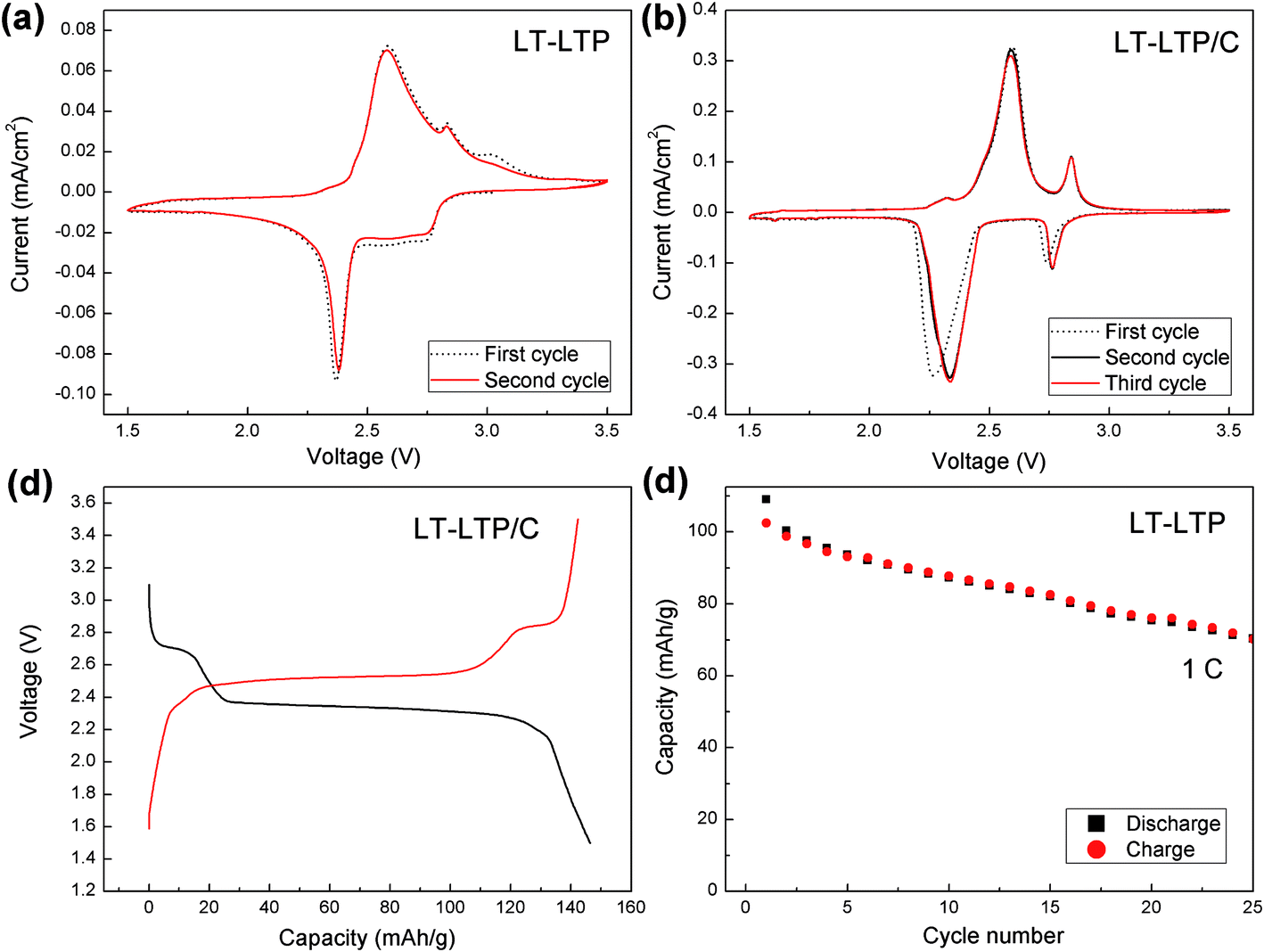 | ||
| Fig. 6 (a) Slow sweep cyclic voltammograms collected from LT-LTP. (b) Slow sweep cyclic voltammograms collected from LT-LTP/C; sweep rate is 0.1 mV s−1. (c) Typical discharge–charge profile of LT-LTP/C (1 C discharge–charge rate). (d) Specific capacity as a function of cycle number for LT-LTP at 1 C discharge–charge rate. | ||
LT-LTP showed a poor cycling stability due to a lack of electronic conductivity. Fig. 6d shows that the material retains a capacity <70 mA h g−1 after 25 cycles at 1 C (140 mA g−1) charge–discharge rate. On the other hand, LT-LTP/C showed an enhanced cycling stability over hundreds of cycles at charge–discharge rates ranging from 1 C (140 mA g−1) to 30 C (4200 mA g−1) at room temperature (Fig. 7a). The material delivered initial discharge capacities of 140 mA h g−1 at 1 C and 110 mA h g−1 at 10 C rate, and retained a capacity 104 mA h g−1 after 800 cycles at 10 C charge–discharge rate (Fig. 7b). A capacity fade is observed in the first 10–15 cycles, followed by an increase of the capacity. The increase of the capacity can be attributed to an enhancement of electrode/electrolyte contact generated upon cycling. LT-LTP/C hence shows a superior performance compared to that reported for many conventional LTP/C composites.8,10,11,13 Only few studies have presented materials with comparable enhanced performance, namely the materials prepared by the groups of Xia2 and Kim.9 However, both groups have used more complicated procedures to coat/composite LTP with carbon. Xia et al.2 have prepared LTP by a sol–gel technique followed by calcination step at 900 °C in N2 atmosphere, and used the chemical vapor deposition (CVD) technique for carbon-coating. LT-LTP/C prepared in this study, however, shows a more porous structure, smaller particle-size, better high-rate performance and better cycling stability than CVD carbon-coated LTP. The group of Kim,9 on the other hand, has presented a good example for an enhanced performance of LTP that is directly related to compositing the material with carbon. In this work, Kim et al. have prepared complex graphene oxide–LTP hybrids which have shown excellent high-rate performance. However, this performance is also comparable to that observed for our simply-synthesized LT-LTP/C. The enhanced performance of LT-LTP/C is clearly related to the improved microstructure and, possibly, the modified electrochemical behavior of LT-LTP. It is not likely that the enhanced performance of LT-LTP/C is solely related to the procedure used to composite LT-LTP with carbon because the same procedure is used by Madhavi et al. yielding LTP/C phases with less promising performance.8,13
 | ||
| Fig. 7 (a) Specific capacity as a function of cycle number for LT-LTP/C at different discharge–charge rates. (b) Specific capacity as a function of cycle number for LT-LTP/C at 10 C discharge–charge rate. | ||
Hence, we attribute the enhanced performance of LT-LTP/C to the modified structural, microstructural and electrochemical properties of the material. A macroporous material has the advantages of providing short lithium diffusion distances in the solid and having better contact with the electrolyte. Our synthesized LT-LTP/C composite combines these advantages with: (1) an improved electronic conductivity due to intimate contact between (interconnected) LTP nanoparticles and nanostructured carbon; (2) ease of synthesis using a template-free procedure, low calcination temperature, and a simple procedure to composite the material with carbon (with no ball milling step). It is also interesting that the employed synthesis procedure has influenced the structural and electrochemical properties of LTP. Our results indicate that about one-third of the M1 sites in the LT-LTP structure are empty which provides a new lithium intercalation regime (two-step intercalation regime) that seems to enhance lithium-exchange with the bulk material. This effect is currently under further investigation.
4. Conclusions
A simple sol–gel procedure with a final calcination step at 550 °C is used to synthesis a low-temperature LTP phase (LT-LTP). The material is macroporous with an average particle size of 50–100 nm. In addition to modifying the microstructure, the employed synthesis procedure has influenced both structural and electrochemical properties of the material. About one third of the M1 sites in the LT-LTP structure are found to be empty, and hence the material exhibits a two-step lithium-exchange regime. LT-LTP/C shows a remarkably enhanced cycle performance compared to conventional LTP/C composites. A simple synthesis and superior cycle performance of LT-LTP/C suggest that the material may replace conventional LTP/C composites in different lithium battery applications.Acknowledgements
The authors acknowledge support by the Humboldt foundation (H. E.-S.), the LOEWE project STORE-E and the LaMa (Laboratory of Materials Research) at Justus-Liebig University (State of Hessen).References
- H. Wang, K. Huang, Y. Zeng, S. Yang and L. Chen, Electrochim. Acta, 2007, 52, 3280–3285 CrossRef CAS.
- J.-Y. Luo and Y.-Y. Xia, Adv. Funct. Mater., 2007, 17, 3877–3884 CrossRef CAS.
- C. Wessells, F. L. Mantia, H. Deshazer, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 158, A352–A355 CrossRef CAS.
- C. Wessells, R. A. Huggins and Y. Cui, J. Power Sources, 2011, 196, 2884–2888 CrossRef CAS.
- R. B. Shivashankaraiah, H. Manjunatha, K. C. Mahesh, G. S. Suresh and T. V. Venkatesha, J. Electrochem. Soc., 2012, 159, A1074–A1082 CrossRef CAS.
- Y. Cui, Y. Hao, W. Bao, Y. Shi, Q. Zhuang and Y. Qiang, J. Electrochem. Soc., 2013, 160, A53–A59 CrossRef CAS.
- J.-Y. Luo, J.-L. Liu, P. He and Y.-Y. Xia, Electrochim. Acta, 2008, 53, 8128–8133 CrossRef CAS.
- V. Aravindan, W. Chuiling, M. V. Reddy, G. V. Subba Rao, B. V. R. Chowdari and S. Madhavi, Phys. Chem. Chem. Phys., 2012, 14, 5808–5814 RSC.
- C. H. Lim, A. G. Kannan, H.-W. Lee and D. K. Kim, J. Mater. Chem. A, 2013, 1, 6183–6190 CAS.
- M. Zhou, L. Liu, L. Yi, Z. Yang, S. Mao, Y. Zhou, T. Hu, Y. Yang, B. Shen and X. Wang, J. Power Sources, 2013, 234, 292–301 CrossRef CAS.
- L. Liu, M. Zhou, W. Wang, H. Guo, F. Tian and X. Wang, Electrochim. Acta, 2012, 70, 136–141 CrossRef CAS.
- C. R. Mariappan, C. Galven, M.-P. Crosnier-Lopez, F. Le Berre and O. Bohnke, J. Solid State Chem., 2006, 179, 450–456 CrossRef CAS.
- V. Aravindan, W. Chuiling and S. Madhavi, RSC Adv., 2012, 2, 7534–7539 RSC.
- C. Delmas, A. Nadiri and J. L. Soubeyroux, Solid State Ionics, 1988, 28–30, 419–423 CrossRef.
- A. Aatiq, M. Menetrier, L. Croguennec, E. Suard and C. Delmas, J. Mater. Chem., 2002, 12, 2971–2978 RSC.
- A. C. Larson and R. B. Von Dreele, General Structural Analysis System, Los Alamos National Laboratory, Los Alamos, NM, 1994 Search PubMed.
- C. Vidal-Abarca, P. Lavela, M. J. Aragon, N. Plylahan and J. L. Tirado, J. Mater. Chem., 2012, 22, 21602–21607 RSC.
- M. Catti, A. Comotti, S. D. Blas and R. M. Ibberson, J. Mater. Chem., 2004, 14, 835–839 RSC.
- A. Aatiq, M. Menetrier, A. El Jazouli and C. Delmas, Solid State Ionics, 2002, 150, 391–405 CrossRef CAS.
- A. Aatiq, C. Delmas and A. El Jazouli, J. Solid State Chem., 2001, 158, 169–174 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |