
Chiral phosphine-phosphoramidite ligands for highly enantioselective hydrogenation of N-arylimines†
Qing Liab,
Chuan-Jin Hou*ab,
Xiao-Ning Liuab,
De-Zhi Huang*a,
Yan-Jun Liua,
Rui-Feng Yanga and
Xiang-Ping Hu*b
aSchool of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian 116034, China. E-mail: houcj@dlpu.edu.cn; huangjj6688@163.com
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xiangping@dicp.ac.cn
First published on 20th January 2015
Abstract
The asymmetric hydrogenation of N-arylimines with the chiral phosphine-phosphoramidite ligand, (Sc,Sa)-PEAPhos 2b, has been developed. The results revealed that the presence of the substituents on the 3,3′-positions of the binaphthyl backbone significantly improved the enantioselectivity. The utility of this methodology was demonstrated in the synthesis of the chiral fungicide (R)-metalaxyl.
Introduction
Chiral amines are important synthetic intermediates in the synthesis of many biologically active natural and unnatural products. As a result, numerous methods are available for their preparation. Among them, catalytic asymmetric hydrogenation of imines has proven to be one of the most direct and convenient routes to chiral amines and their derivatives, due to its inherent efficiency and atom economy.1 Although great efforts have been made during the past two decades, the development of highly efficient catalytic asymmetric hydrogenation of imines remains a challenging task, in contrast to the relative maturity of catalytic asymmetric hydrogenation of olefins and ketones, probably due to the interconversion between E/Z isomers of imines and the poisoning effect of the resulting amines on the catalyst. In 1975, Scorrano and co-workers2 reported the first catalytic asymmetric hydrogenation of N-benzylimine with an Rh-diphosphine catalyst. Since then, many highly enantioselective chiral catalysts have been developed and a variety of imine frameworks including cyclic and acyclic imines have been efficiently hydrogenated in high enantioselectivities. Except for the well-known iridium-Xyliphos catalyst3 applied to the industrial production of chiral herbicide (S)-Metolachlor, Buchwald's titanocene catalyst,4 Pfaltz's iridium-PHOX catalyst,5 Zhang's iridium-f-Binaphane catalyst,6 Zhou's iridium-SIPHOX catalyst7 also exhibited excellent results for asymmetric hydrogenation of various imines. Despite these impressive achievements,8 asymmetric hydrogenation of imines still has challenges of low reactivity, narrow substrate scope and harsh reaction conditions. Therefore, development of a new catalyst system with high reactivity and enantioselectivity as well as broad substrate scope for asymmetric hydrogenation of imines is still highly desirable.Recently, we and other groups developed a series of unsymmetrical hybrid chiral phosphine-phosphoramidite ligands. These ligands have the advantages of easy accessibility, modularity and stability toward air and moisture, which can be exposed to air for several months without any changes in their 1H or 31P NMR spectra and any loss in the catalytic activity and enantioselectivity. Therefore, these ligands have been found to display wide utility in asymmetric catalysis.9–15 However, there are few reports on the asymmetric hydrogenation of imines with chiral phosphine-phosphoramidite ligands. The only one example was reported by us recently,16 in which a new H8-BINOL-derived phosphine-phosphoramidite ligand, (Rc,Ra)-1, was found to be highly efficient in the enantioselective hydrogenation of sterically hindered N-arylimines, a class of challenging substrates. Further research disclosed that the steric effect of substituents on the 3,3′-postions of binaphthyl backbone of the ligand showed a significant influence on the enantioselectivity of the asymmetric hydrogenation of N-arylimines. As a result, we report herein the asymmetric hydrogenation of N-arylimines with a 3,3′-disubstituted binaphthyl ligand, (Sc,Sa)-PEAPhos 2, in which high turnover numbers (up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and excellent enantioselectivity (up to 98% ee) were obtained (Fig. 1).
000) and excellent enantioselectivity (up to 98% ee) were obtained (Fig. 1).
 | ||
| Fig. 1 Chiral phosphine-phosphoramidite ligands, (Rc,Ra)-1 and (Sc,Sa)-PEAPhos 2. | ||
Results and discussion
The chiral phosphine-phosphoramidite ligand (Sc,Sa)-PEAPhos 2a–c were synthesized from commercially available (S)-1-phenylethylamine as we have reported previously.10e,u With these ligands in hand, we then examined their efficiency in the iridium-catalyzed asymmetric hydrogenation of N-arylimines. N-(1-Phenylethylidene)benzenamine 3a was selected as a model substrate for the screening process and the result are summarized in Table 1. The initial hydrogenation was carried out in the presence of 1 mol% of catalyst prepared in situ from [Ir(COD)Cl]2 and 2.2 equiv. of chiral ligand with KI as an additive. To our delight, (Sc,Sa)-PEAPhos 2a displayed a promising performance in this reaction, affording N-(1-phenylethyl)benzenamine 4a in full conversion with 80% ee (entry 1). Since the steric hindrance of a chiral ligand normally affects the catalytic activity and enantioselectivity, we then decided to investigate the ligands 2b–c with more sterically hindered substituents on the 3,3′-postions of binaphthyl backbone. As expected, ligand 2b with two methyl group on 3,3′-postions provided an ee value of 93%, remarkably superior to that obtained with the parent ligand 2a (entry 2). However, replacing methyl groups with more sterically hindered phenyl groups on the 3,3′-postions, the enantioselectivity decreased significantly (entry 3). The solvent effect was next investigated with ligand 2b. It was found that all of CH2Cl2, toluene and THF were suitable solvents for this reaction, with CH2Cl2 being optimal in terms of conversion and enantioselectivity (entries 4–7).
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Conv.b (%) | eec (%) |
| a Hydrogenation was carried out with 5 mol% of KI and 1 mol% of Ir-catalyst at rt for 24 h.b Determined by GC.c Determined by HPLC using a chiral stationary phase. | ||||
| 1 | 2a | CH2Cl2 | >99 | 80 |
| 2 | 2b | CH2Cl2 | >99 | 93 |
| 3 | 2c | CH2Cl2 | >99 | 29 |
| 4 | 2b | Toluene | >99 | 90 |
| 5 | 2b | THF | >99 | 90 |
| 6 | 2b | AcOEt | >99 | 86 |
| 7 | 2b | MeOH | >99 | 62 |
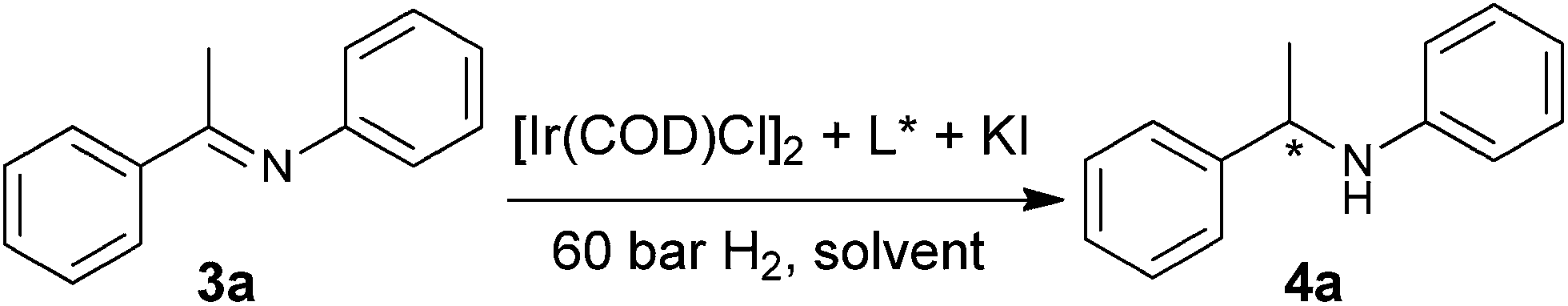
Under the optimal reaction conditions, a range of N-arylimines were then investigated. As can be seen from Table 2, a wide spectrum of N-arylimines with electron-donating and -withdrawing substituents on the phenyl ring were hydrogenated with full conversions and excellent enantioselectivities (entries 1–7). The highest enantioselectivity (97% ee) was achieved in the hydrogenation of N-(1-(3-nitrophenyl)ethylidene)benzene-amine 3e (entry 5). Notably, the hydrogenation appeared to be sensitive to the steric property of the substituents on the aromatic ring. For substrates 3h–j having a o-substituent on the phenyl ring, a dramatic decrease of enantioselectivities (62–78% ee) was observed (entries 8–11). Hydrogenation of 2-naphthyl substrate 3k also gave good ee-value (entry 12). Meanwhile, a heteroaromatic derivative 3l was also suitable for this hydrogenation (entry 13). These results suggested that a variety of N-arylimines, which bear substituted groups such as methoxy, nitro, fluoro, chloro in the phenyl ring, as well as naphthyl and heteroaromatic derivatives, all could be hydrogenated to afford the corresponding chiral amines in good to excellent enantioselectivities.
|
|||
|---|---|---|---|
| Entry | 3 (R, Ar) | Yieldb (%) | eec (%) |
| a Hydrogenation was carried out with 5.0 mol% of KI and 1.0 mol% of Ir-catalyst in 2 mL CH2Cl2, 60 bar of H2, at rt for 24 h, unless otherwise stated.b Yields of isolated product.c Enantiomeric excesses were determined by chiral HPLC.d Using 2c as ligand.e Using 2a as ligand.f Using I2 as additive instead of KI. | |||
| 1 | 3a (R = H, Ar = C6H5) | 99 | 93 |
| 2 | 3b (R = H, Ar = 4-CH3OC6H4) | 99 | 92 |
| 3 | 3c (R = H, Ar = 4-ClC6H4) | 98 | 94 |
| 4 | 3d (R = H, Ar = 4-NO2C6H4) | 98 | 93 |
| 5 | 3e (R = H, Ar = 3-NO2C6H4) | 98 | 97 |
| 6 | 3f (R = H, Ar = 3,4-ClC6H3) | 95 | 93 |
| 7 | 3g (R = 4-F, Ar = C6H5) | 95 | 95 |
| 8d | 3h (R = H, Ar = 2-CH3C6H4) | 95 | 62 |
| 9e | 3i (R = 2-CH3, Ar = C6H5) | 95 | 72 |
| 10f | 3j (R = 2-CF3, Ar = C6H5) | 92 | 78 |
| 11 | 3k (R = H, Ar = 6-CH3O-2-naphthyl) | 98 | 83 |
| 12 | 3l (R = H, Ar = thienyl) | 99 | 85 |

Encouraged by these promising results, we then investigated the hydrogenation of more challenging, sterically hindered N-arylimines 5. The results are summarized in Table 3. The additive was investigated because of its significant influence on the asymmetric catalysis.17 It revealed that the use of I additives could significantly increase the enantioselectivity, which was in accordance with the result reported before by us.16 I2 and KI gave the similar results, while I2 was slightly superior to KI with respect to enantioselectivity in the hydrogenation of sterically hindered N-arylimines (entries 1 and 2). Under the optimized condition, a variety of sterically hindered N-arylalkylarylimines and N-aryldialkylimines could be hydrogenated in good to excellent enantioselectivities. For N-arylarylalkylimines 5a–f, the hydrogenation proceeded smoothly and afforded the corresponding sterically hindered chiral amines in excellent enantioselectivities (94–98% ee) (entries 1–7). The electronic property of substituents on the phenyl ring showed no apparent influence on the enantioselectivity. For the hydrogenation of 2-naphthyl substrate 5g, a slight decrease of enantioselectivity (92% ee) was obtained (entry 8). It is noteworthy that the hydrogenation of more challenging N-aryldialkylimines 5h and 5i were conducted smoothly with good enantioselectivities (84% and 92% ee, respectively) (entries 9 and 10). These results demonstrated the high efficiency of the present catalytic system in the enantioselective hydrogenation of a broad scope of sterically hindered N-arylimines.
|
|||
|---|---|---|---|
| Entry | Imine (R) | Yieldb (%) | eec (%) |
| a Hydrogenation was carried out with 5.0 mol% of I2 and 1.0 mol% of Ir-catalyst in 2 mL CH2Cl2, 60 bar of H2, at rt for 24 h, unless otherwise stated.b Yields of isolated product.c Enantiomeric excesses were determined by chiral HPLC or by chiral GC.d Using KI as additive.e Hydrogenation was performed at 60 °C under a H2 pressure of 70 bar. | |||
| 1 | 5a: R = C6H5 | 98 | 96 |
| 2d | 5a: R = C6H5 | 98 | 94 |
| 3 | 5b: R = 4-CH3C6H4 | 98 | 94 |
| 4 | 5c: R = 4-BrC6H4 | 98 | 96 |
| 5 | 5d: R = 4-NO2C6H4 | 99 | 98 |
| 6 | 5e: R = 3-NO2C6H4 | 98 | 98 |
| 7 | 5f: R = 3-CH3OC6H4 | 98 | 95 |
| 8 | 5g: R = 6-CH3O-2-naphthyl | 92 | 92 |
| 9e | 5h: R = i-Pr | 97 | 84 |
| 10 | 5i: R = MeOCH2 | 95 | 92 |
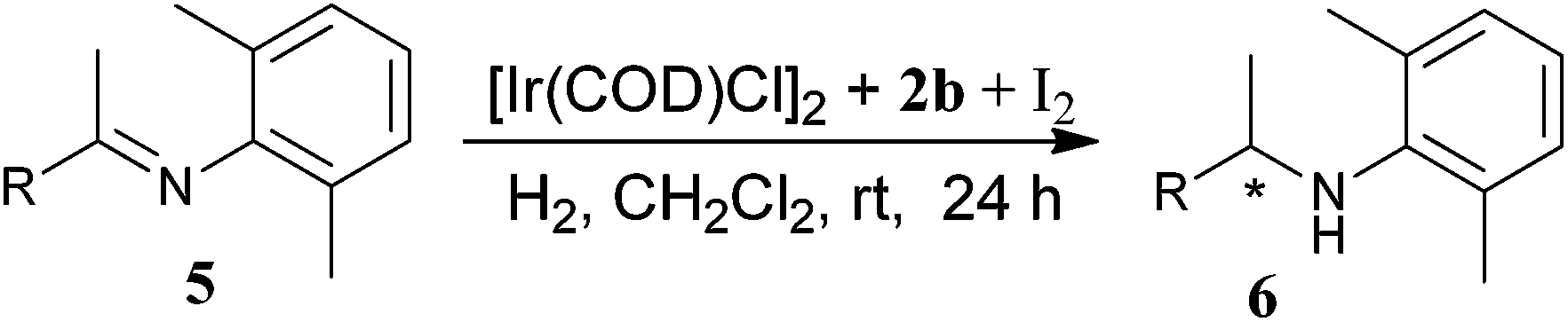
To explore the synthetic utility of the current method, we attempted this methodology as the key step in the enantioselective synthesis of the chiral fungicide (R)-metalaxyl as shown in Scheme 1. With the present catalytic system, hydrogenation of imine 5j afforded the corresponding chiral amine 6j in 95% ee with full conversion, even when the catalyst loading was lowered to 0.002 mol% (S/C = 50000/1). It should be noted that the asymmetric hydrogenation of 5j can be carried out on a gram scale and operated in air without loss of the enantioselectivity and activity. The resulting product 6j can easily be converted to herbicide (R)-metalaxyl 7,18 demonstrating the potential utility of this method in the synthesis of chiral pesticides.
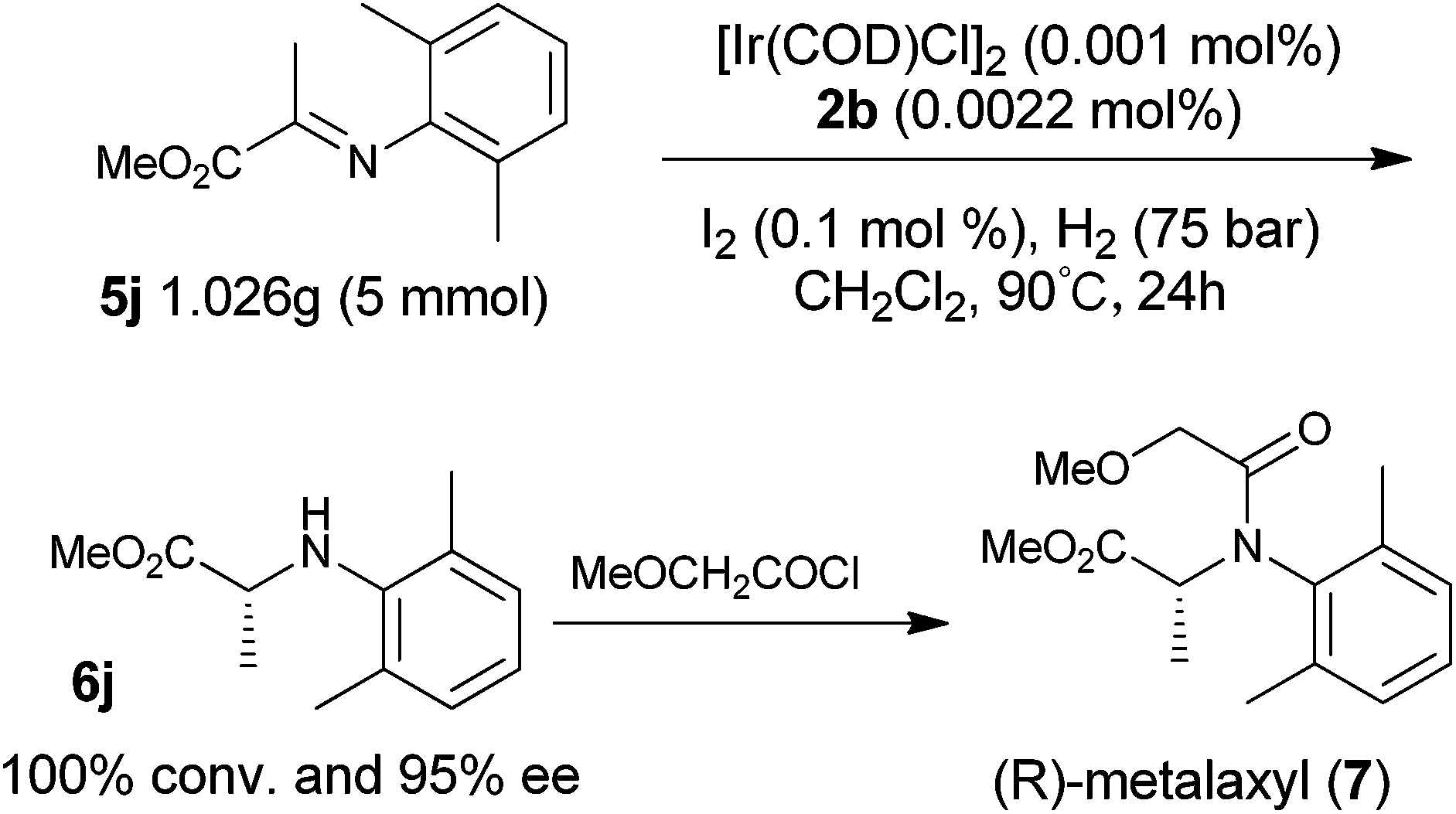 | ||
| Scheme 1 Synthesis of chiral fungicide (R)-metalaxyl on gram scale. | ||
Conclusions
In conclusion, we have demonstrated the asymmetric hydrogenation of N-arylimines with chiral phosphine-phosphoramidite ligand, (Sc,Sa)-PEAPhos 2b, in which high turnover numbers (up to 50000) and excellent enantioselectivity (up to 98% ee) were achieved. The results revealed that the presence of the substituents on the 3,3′-positions of the binaphthyl backbone significantly improved the enantioselectivity. The utility of this methodology was demonstrated by the synthesis of chiral fungicide (R)-metalaxyl at a catalyst loading of 0.002 mol%. Further applications of chiral phosphine-phosphoramidite ligand in asymmetric catalysis are in progress.
Experimental section
General method
All reactions were carried out under a nitrogen atmosphere. Solvents were purified by standard procedure before use. Commercial reagents were used without further purification. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer. Chemical shifts for protons are reported in parts per million (ppm) downfield from tetramethylsilane (TMS) and are referenced to residual protium in the NMR solvent (CHCl3 = δ 7.26). Enantiomeric ratios were determined by chiral HPLC with n-hexane and i-PrOH as solvents.General procedure for the synthesis of phosphine-phosphoramidite ligands 2a–2c
Chiral phosphine-phosphoramidite ligands were synthesized according to literature.10e,u To a stirred solution of (Sa)-chlorophosphite (1.0 mmol) in dried toluene (4.0 mL) at 0 °C was added a solution of (Sc)-DPPNHMe (1.0 mmol) and Et3N (3.0 mmol) in dried toluene (4.0 mL) within 30 min. The resulting mixture was stirred overnight at room temperature. The precipitate was filtered, and the solid was washed with toluene. The filtrate was collected, and concentrated under reduced pressure to give the crude product which was further purified by column chromatography.General procedure for asymmetric hydrogenation
In a nitrogen-filled glovebox, a stainless steel autoclave was charged with [Ir(COD)Cl]2 (0.0025 mmol), (Sc,Sa)-2b (0.0055 mmol) and KI or I2 (0.025 mmol) in 1.0 mL of a degassed CH2Cl2. After stirring for 10 min at room temperature, a solution of the imine substrates 3 or 5 (0.5 mmol) in 1.0 mL of CH2Cl2 was added to the reaction mixture, and then the hydrogenation was performed at room temperature under an H2 pressure of 60 bar for 24 hours. The solvent was then evaporated and the residue was purified by flash column chromatography to give the corresponding hydrogenation product 4 or 6, which was analysed by chiral GC or chiral HPLC to determine the enantiomeric excesses.Acknowledgements
The authors are grateful for the financial supports from the National Natural Science Foundation of China (21403022) and Program for Liaoning Excellent Talents in University (LJQ2013059).Notes and references
- For some recent reviews on asymmetric hydrogenation of imines, see (a) N. Fleury-Bregeot, V. de la Fuente, S. Castillon and C. Claver, ChemCatChem, 2010, 2, 1346 CrossRef CAS; (b) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753 CrossRef CAS; (c) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (d) D. Wang, C.-J. Hou, L.-F. Chen, X.-N. Liu, Q.-D. An and X.-P. Hu, Chin. J. Org. Chem., 2013, 33, 1355 CrossRef CAS; (e) A. Bartoszewicz, N. Ahlsten and B. Martín-Matute, Chem.–Eur. J., 2013, 19, 7274 CrossRef CAS PubMed.
- A. Levi, G. Modena and G. Scorrano, J. Chem. Soc., Chem. Commun., 1975, 6 RSC.
- (a) H.-U. Blaser, H.-P. Buser, K. Loers, R. Hanreich, H. P. Jalett, E. Jelsch, H. D. Schneider, F. Spindler and A. Wagmann, Chimia, 1999, 53, 275 CAS; (b) H.-U. Blaser, C. Malan, B. Pugin, F. Spinder, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103 CrossRef CAS; (c) H.-U. Blaser, B. Pugin, F. Spindler and M. Thommen, Acc. Chem. Res., 2007, 40, 1240 CrossRef CAS PubMed.
- (a) C. A. Willoughbly and S. L. Buchwald, J. Am. Chem. Soc., 1992, 114, 7562 CrossRef; (b) C. A. Willoughbly and S. L. Buchwald, J. Org. Chem., 1993, 58, 7627 CrossRef; (c) C. A. Willoughbly and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 8952 CrossRef; (d) C. A. Willoughbly and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 11703 CrossRef.
- (a) P. Schbider, G. Koch, R. Pretot, G. Wang, F. M. Bohnen, C. Kruger and A. Pfaltz, Chem.–Eur. J., 1997, 3, 887 CrossRef; (b) A. Baeza and A. Pfaltz, Chem.–Eur. J., 2010, 16, 4003 CrossRef CAS PubMed; (c) A. Franzke, F. Voss and A. Pfaltz, Tetrahedron, 2011, 67, 4358 CrossRef CAS PubMed; (d) S. Kainz, A. Brinkmann, W. Leitner and A. Pfaltz, J. Am. Chem. Soc., 1999, 121, 6421 CrossRef CAS; (e) M. Solinas, A. Pfaltz, P. G. Cozzi and W. Leitner, J. Am. Chem. Soc., 2004, 126, 16142 CrossRef CAS PubMed; (f) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS PubMed.
- (a) D. Xiao and X. Zhang, Angew. Chem., Int. Ed., 2001, 40, 3425 CrossRef CAS; (b) G. Hou, F. Gosselin, W. Li, J. C. Mcwillams, Y. Sun, M. Weisel, P. D. O'shea, C. Chen, I. W. Davies and X. Zhang, J. Am. Chem. Soc., 2009, 131, 9882 CrossRef CAS PubMed; (c) M. Chang, W. Li, G. Hou and X. Zhang, Adv. Synth. Catal., 2010, 352, 3121 CrossRef CAS; (d) M. Chang, W. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10679 CrossRef CAS PubMed.
- (a) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS PubMed; (b) J.-H. Xie, P.-C. Yan, Q.-Q. Zhang, K.-X. Yuan and Q.-L. Zhou, ACS Catal., 2012, 2, 561 CrossRef CAS.
- For selected examples of Ir-catalyzed asymmetric hydrogenation of imines, see: (a) X.-B. Jiang, A. J. Minnaard, B. Hessen, B. L. Feringa, A. L. L. Duchateau, J. G. O. Andrien, J. A. Boogers and J. G. de Vries, Org. Lett., 2003, 5, 1503 CrossRef CAS PubMed; (b) E. Guiu, B. Munoz, S. Castillon and C. Claver, Adv. Synth. Catal., 2003, 345, 169 CrossRef CAS; (c) C. Moessner and C. Bolm, Angew. Chem., Int. Ed., 2005, 44, 7564 CrossRef CAS PubMed; (d) T. Imamoto, N. Iwadate and K. Yoshida, Org. Lett., 2006, 8, 2289 CrossRef CAS PubMed; (e) S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef CAS PubMed; (f) M. T. Reetz and O. Bondarev, Angew. Chem., Int. Ed., 2007, 46, 4523 CrossRef CAS PubMed; (g) C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2008, 130, 14450 CrossRef CAS PubMed; (h) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967 CrossRef CAS PubMed; (i) C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 130, 14450 CrossRef PubMed; (j) S. Shirai, H. Nara, Y. Kayaki and T. Ikariya, Organometallics, 2009, 28, 802 CrossRef CAS; (k) Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345 CrossRef CAS PubMed; (l) W. Li, G. Hou, M. Chang and X. Zhang, Adv. Synth. Catal., 2009, 351, 3123 CrossRef CAS; (m) N. Mrsic, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2009, 131, 8358 CrossRef CAS PubMed; (n) G. Hou, F. Gosselin, W. Li, J. C. McWilliams, Y. Sun, M. Weisel, P. D. O'Shea, C.-Y. Chen, I. W. Davies and X. Zhang, J. Am. Chem. Soc., 2009, 131, 9882 CrossRef CAS PubMed; (o) A. Beaza and A. Pfaltz, Chem.–Eur. J., 2010, 16, 4003 CrossRef PubMed; (p) G. Hou, R. Tao, Y. Sun, X. Zhang and F. Gosselin, J. Am. Chem. Soc., 2010, 132, 2124 CrossRef CAS PubMed; (q) K. Gao, C.-B. Yu, W. Li, Y.-G. Zhou and X. Zhang, Chem. Commun., 2011, 47, 7845 RSC; (r) K. Gao, C.-B. Yu, D.-S. Wang and Y.-G. Zhou, Adv. Synth. Catal., 2012, 354, 483 CrossRef CAS; (s) K. Gao, B. Wu, C.-B. Yu, Q.-A. Chen, Z.-S. Ye and Y.-G. Zhou, Org. Lett., 2012, 14, 3890 CrossRef CAS PubMed; (t) J.-H. Xie, P.-C. Yan, Q.-Q. Zhang, K.-X. Yuan and Q.-L. Zhou, ACS Catal., 2012, 2, 561 CrossRef CAS; (u) R.-N. Guo, K. Gao, Z.-S. Ye, L. Shi, Y. Li and Y.-G. Zhou, Pure Appl. Chem., 2013, 85, 843 CrossRef CAS; (v) B. Ma, Z. Ding, J. Liu, Y. He and Q.-H. Fan, Chem.–Asian J., 2013, 8, 1101 CrossRef CAS PubMed; (w) W. Tang and J. Xiao, Synthesis, 2014, 46, 1297 CrossRef PubMed.
- For reviews, see: (a) L. Eberhardt, D. Armspach, J. Harrowfield and D. Matt, Chem. Soc. Rev., 2008, 37, 839 RSC; (b) F. Boeda, T. Beneyton and C. Crevisy, Mini-Rev. Org. Chem., 2008, 5, 96 CrossRef CAS; (c) J. Wassenaar and J. N. H. Reek, Org. Biomol. Chem., 2011, 9, 1704 RSC.
- For Rh-catalyzed asymmetric reactions, see: (a) G. Franciò, F. Faraone and W. Leitner, Angew. Chem., Int. Ed., 2000, 39, 1428 CrossRef; (b) X. Jia, X. Li, W. S. Lam, S. H. L. Kok, L. Xu, G. Lu, C.-H. Yeung and A. S. C. Chan, Tetrahedron: Asymmetry, 2004, 15, 2273 CrossRef CAS PubMed; (c) X.-P. Hu and Z. Zheng, Org. Lett., 2004, 6, 3585 CrossRef CAS PubMed; (d) X.-P. Hu and Z. Zheng, Org. Lett., 2005, 7, 419 CrossRef CAS PubMed; (e) J.-D. Huang, X.-P. Hu, Z.-C. Duan, Q.-H. Zeng, S.-B. Yu, J. Deng, D.-Y. Wang and Z. Zheng, Org. Lett., 2006, 8, 4367 CrossRef CAS PubMed; (f) K. A. Vallianatou, I. D. Kostas, J. Holz and A. Boner, Tetrahedron Lett., 2006, 47, 7947 CrossRef CAS PubMed; (g) W. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2006, 45, 5515 CrossRef CAS PubMed; (h) W. Zhang and X. Zhang, J. Org. Chem., 2007, 72, 1020 CrossRef CAS PubMed; (i) Y. Yan and X. Zhang, J. Am. Chem. Soc., 2006, 128, 7198 CrossRef CAS PubMed; (j) D.-Y. Wang, X.-P. Hu, J.-D. Huang, J. Deng, S.-B. Yu, Z.-C. Duan, X.-F. Xu and Z. Zheng, Angew. Chem., Int. Ed., 2007, 46, 7810 CrossRef CAS PubMed; (k) J. Wassenaar, M. Kuil and J. N. H. Reek, Adv. Synth. Catal., 2008, 350, 1610 CrossRef CAS; (l) M. Qiu, X.-P. Hu, D.-Y. Wang, J. Deng, J.-D. Huang, S.-B. Yu, Z.-C. Duan and Z. Zheng, Adv. Synth. Catal., 2008, 350, 1413 CrossRef CAS; (m) S.-B. Yu, J.-D. Huang, D.-Y. Wang, X.-P. Hu, J. Deng, Z.-C. Duan and Z. Zheng, Tetrahedron: Asymmetry, 2008, 19, 1862 CrossRef CAS PubMed; (n) M. Qiu, D.-Y. Wang, X.-P. Hu, J.-D. Huang, S.-B. Yu, J. Deng, Z.-C. Duan and Z. Zheng, Tetrahedron: Asymmetry, 2009, 20, 210 CrossRef CAS PubMed; (o) J. Wassenaar and J. N. H. Reek, J. Org. Chem., 2009, 74, 8403 CrossRef CAS PubMed; (p) J. Wassenaar, M. Kuil, M. Lutz, A. L. Spek and J. N. H. Reek, Chem.–Eur. J., 2010, 16, 6509 CrossRef CAS PubMed; (q) J. Wassenaar, B. de Bruin and J. N. H. Reek, Organometallics, 2010, 29, 2767 CrossRef CAS; (r) T. Pullmann, B. Engendahl, Z. Zhang, M. Holscher, A. Zanotti-Gerosa, A. Dyke, G. Francio and W. Leitner, Chem.–Eur. J., 2010, 16, 7517 CrossRef CAS PubMed; (s) U. Hintermair, T. Hofener, T. Pullmann, G. Francio and W. Leitner, ChemCatChem, 2010, 2, 150 CrossRef CAS; (t) X. Zhang, B. Cao, Y. Yan, S. Yu, B. Ji and X. Zhang, Chem.–Eur. J., 2010, 16, 871 CrossRef CAS PubMed; (u) X.-M. Zhou, J.-D. Huang, L.-B. Luo, C.-L. Zhang, X.-P. Hu and Z. Zheng, Org. Biomol. Chem., 2010, 8, 2320 RSC; (v) X. Zhang, B. Cao, S. Yu and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 4047 CrossRef CAS PubMed; (w) S. H. Chikkali, R. Bellini, B. de Bruin, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 6607 CrossRef CAS PubMed; (x) T. M. Konrad, P. Schmitz, W. Leitner and G. Franciò, Chem.–Eur. J., 2013, 19, 13299 CrossRef CAS PubMed.
- For Ru-catalyzed asymmetric reaction, see: S. Burk, G. Franciò and W. Leitner, Chem. Commun., 2005, 3460 RSC.
- For Ag-catalyzed asymmetric reaction, see: S.-B. Yu, X.-P. Hu, J. Deng, D.-Y. Wang, Z.-C. Duan and Z. Zheng, Tetrahedron: Asymmetry, 2009, 20, 621 CrossRef CAS PubMed.
- For Cu-catalyzed asymmetric reactions, see: (a) F. Boeda, D. Rix, H. Clavier, C. Crevisy and M. Mauduit, Tetrahedron: Asymmetry, 2006, 17, 2726 CrossRef CAS PubMed; (b) C.-J. Hou, W.-L. Guo, X.-P. Hu, J. Deng and Z. Zheng, Tetrahedron: Asymmetry, 2011, 22, 195 CrossRef CAS PubMed.
- For Pd-catalyzed asymmetric reactions, see: (a) J. Wassenaar, S. van Zutphen, G. Mora, P. Le Floch, M. A. Siegler, A. L. Spek and J. N. H. Reek, Organometallics, 2009, 28, 2724 CrossRef CAS; (b) J. Wassenaar, E. Jansen, W.-J. van Zeist, F. M. Bickelhaupt, M. A. Siegler, A. L. Spek and J. N. H. Reek, Nat. Chem., 2010, 2, 417 CrossRef CAS PubMed.
- For Rh-, Ru-, Ir-catalyzed asymmetric hydrogenation, see: M. Eggenstein, A. Thomas, J. Theuerkauf, G. Francio and W. Leitner, Adv. Synth. Catal., 2009, 351, 725 CrossRef CAS.
- C.-J. Hou, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, Org. Lett., 2012, 14, 3554 CrossRef CAS PubMed.
- For some reviews in additive effects on asymmetric catalysis, see: (a) K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26 CrossRef CAS; (b) T. Nagano, A. Iimuro, K. Yamaji, Y. Kita and K. Mashima, Heterocycles, 2014, 88, 103 CrossRef CAS PubMed.
- (a) H.-U. Blaser and F. Spindler, Top. Catal., 1997, 4, 275 CrossRef; (b) J. Boëlle, R. Schneider, P. Gérardin, B. Loubinoux, P. Maienfisch and A. Rindlisbacher, Pestic. Sci., 1998, 54, 302 CrossRef; (c) O.-J. Park, S.-H. Lee, T.-Y. Park, S.-W. Lee and K.-H. Cho, Tetrahedron: Asymmetry, 2005, 16, 1221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra and HPLC for ee analysis. See DOI: 10.1039/c4ra16062b |
| This journal is © The Royal Society of Chemistry 2015 |