DOI:
10.1039/C4RA15905E
(Paper)
RSC Adv., 2015,
5, 14747-14755
Positive and negative allosteric effects of thiacalix[4]arene-based receptors having urea and crown-ether moieties†
Received
6th December 2014
, Accepted 22nd January 2015
First published on 22nd January 2015
Abstract
Heteroditopic receptors (4a–e) based on a thiacalix[4]arene in the 1,3-alternate conformation, which have two urea moieties linking various phenyl groups substituted with either electron-donating or -withdrawing groups at their m-, or p-positions with a crown-ether moiety at the opposite side of the thiacalix[4]arene cavity, have been synthesized. The two examples with p-CH3– (4b) and p-NO2-substituted (4e) phenyl groups have been characterized by X-ray crystallography. The binding properties of receptor 4e were investigated by means of 1H NMR spectroscopic and absorption titration experiments in CHCl3–DMSO (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution in the presence of K+ ions and various anions. Interestingly, it was found that receptor 4e, which possesses two p-nitrophenyl ureido moieties, can complex most efficiently in the urea cavity or the crown-ether moiety; and the plausible allosteric effect of receptor 4e was also studied.
:1, v/v) solution in the presence of K+ ions and various anions. Interestingly, it was found that receptor 4e, which possesses two p-nitrophenyl ureido moieties, can complex most efficiently in the urea cavity or the crown-ether moiety; and the plausible allosteric effect of receptor 4e was also studied.
Introduction
The use of calix[n]arenes1 as building blocks for receptors capable of the highly selective recognition of cations, anions or neutral molecules has received considerable attention in the field of supramolecular chemistry. Among the various kinds of calix[n]arenes available, thiacalix[4]arenes2,3 are proving to be competent scaffolds and are finding wide use, for example as chemosensors, as well as in catalysis because of their favourable conformational properties, easy functionalization and emerging metal coordination chemistry. Several kinds of systems based on thiacalix[4]arenes are suitable for allosteric regulation4 of host–guest interactions with metal cations, and these contribute greatly to organic processes in biological systems. Anions also play an important role in biological processes, and are closely related with biological systems such as DNA and enzyme substrates. The development and the investigation of anion selective sensors5 have attracted considerable interest. However, it is more difficult to accomplish compared with metal cation sensors because anions can possess structures of different shapes,6 typically spherical (F−, Cl−, Br−, I−), Y-shaped (AcO−, PhCOO−) or tetrahedral (H2PO4−). In recent years, anion receptors based on calix[n]arenes have become an active research topic. Calix[n]arene urea derivatives are efficient for anion recognition given the hydrogen-bonding interaction between anions and N–H protons which can occur.
Colorimetric chemosensors7,8 have also attracted attention due to some desirable features such as easy detection by the naked eye, construction of simple, low-cost devices and so on. Many colorimetric anion receptors containing a variety of chromogenic signaling units such as indole, imidazolium, benzenediimide, 4-nitrophenylazo, diazo and anthraquinone groups have been developed. Furthermore, numerous colorimetric anion sensors utilizing a variety of structural scaffolds, which contain urea groups, have been investigated and proved to be efficient naked-eye detectors for various anions. However, there are a few reports on the development of colorimetric chemosensors based calix[4]arene type scaffolds.8l,p
Lhoták9 and co-workers have reported anion receptors based on either an upper rim substituted calix[4]arene or thiacalix[4]-arene, which contains two p-nitrophenyl or p-tolyl ureido moieties.9a–c,h These anion receptors exhibited effective recognition abilities towards selected anions in common organic solvents. Moreover, Kumar10 and co-workers reported an anion receptor bearing a calix[4]arene in the 1,3-alternate conformation, which contains two p-nitrophenyl moieties.10g This compound exhibited strong binding and good selectivity for Cl− ion due to the formation of strong hydrogen bonds between the Cl− ion and N–H protons in common organic solvents. However, investigations concerning the appearance of an allosteric effect in analogues based on the interaction of thiacalix[4]arene and alkali metal cations and anions has not yet been reported.
Herein, we have independently designed a heterodimeric system11 based on a thiacalix[4]arene having two different side arms, viz two ureas moieties linking various phenyl groups bearing either electron-donating or -withdrawing groups at their m-, or p-positions. The calixarene also has a crown ether moiety at the opposite side of the thiacalix[4]arene cavity. We herein put forward the hypothesis (and then demonstrate) that the heterodimeric system, which is controlled by the complexation of the opposing side arms with anions and K+ ion, exhibits effective positive and negative allosteric effects.
Results and discussions
Synthesis
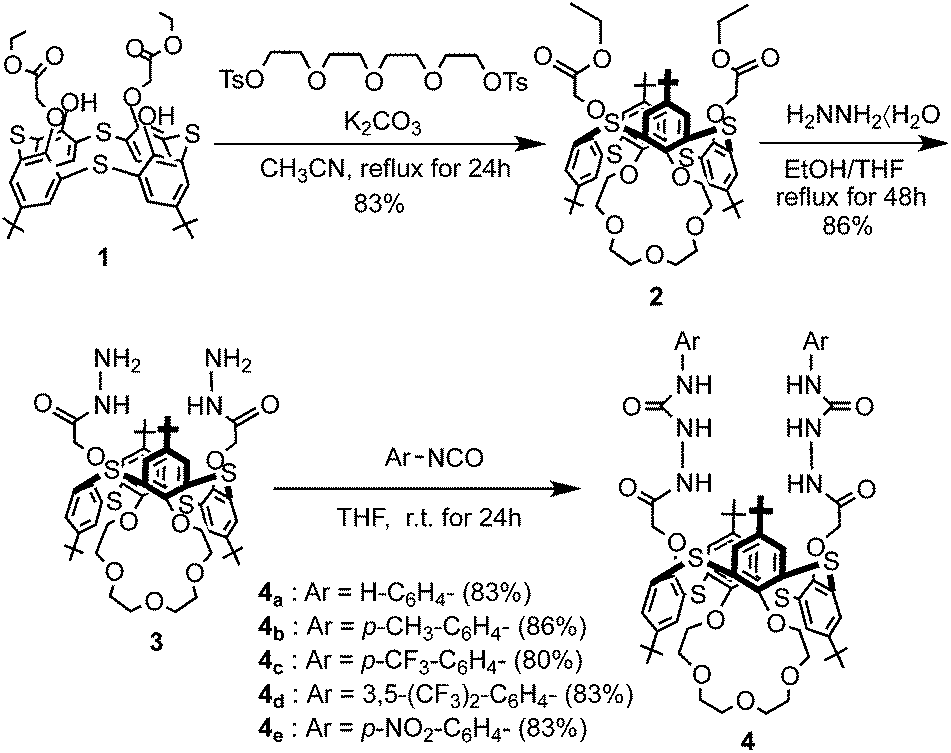
The O-alkylation of distal-1 was carried out with 1.5 equivalents of tetraethyleneglycol ditosylate in the presence of an equivalent of K2CO3 according to the reported procedure, and afforded the desired 1,3-alternate-2 in 83% yield.12 The hydrazinolysis of 1,3-alternate-2 was carried out with a large excess of hydrazine hydrate, and afforded the desired 1,3-alternate-3 in 86% yield. The condensation of 1,3-alternate-3 with 2.2 equivalents of the appropriate isocyanate in THF furnished the receptors 4a–e in good to excellent yields (Scheme 1). In general, the 1H NMR spectrum of receptors 4a–e in CDCl3–DMSO (10:1, v/v) exhibited the characteristics of a 1,3-alternate conformation such as two singlets (18H each) for the tert-butyl protons, one singlet (4H) for OCH2CO protons, two singlets (4H each) for aromatic protons and two singlets (2H each) for four urea NH protons.
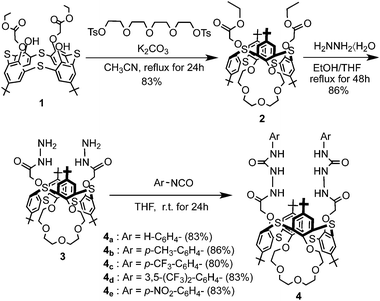 |
| | Scheme 1 Synthesis of receptors 1,3-alternate-4a–e. | |
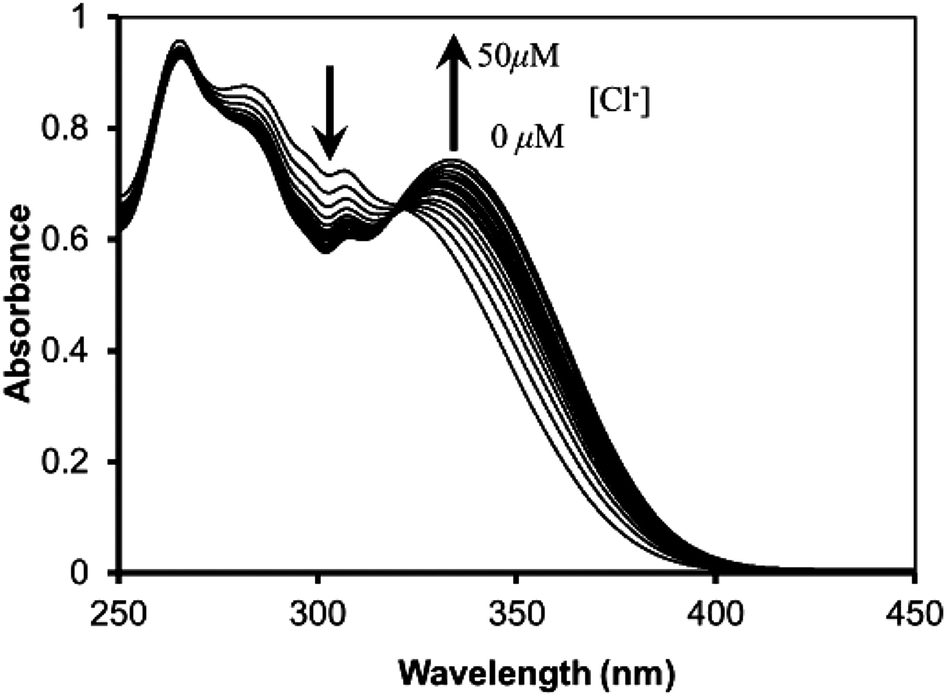
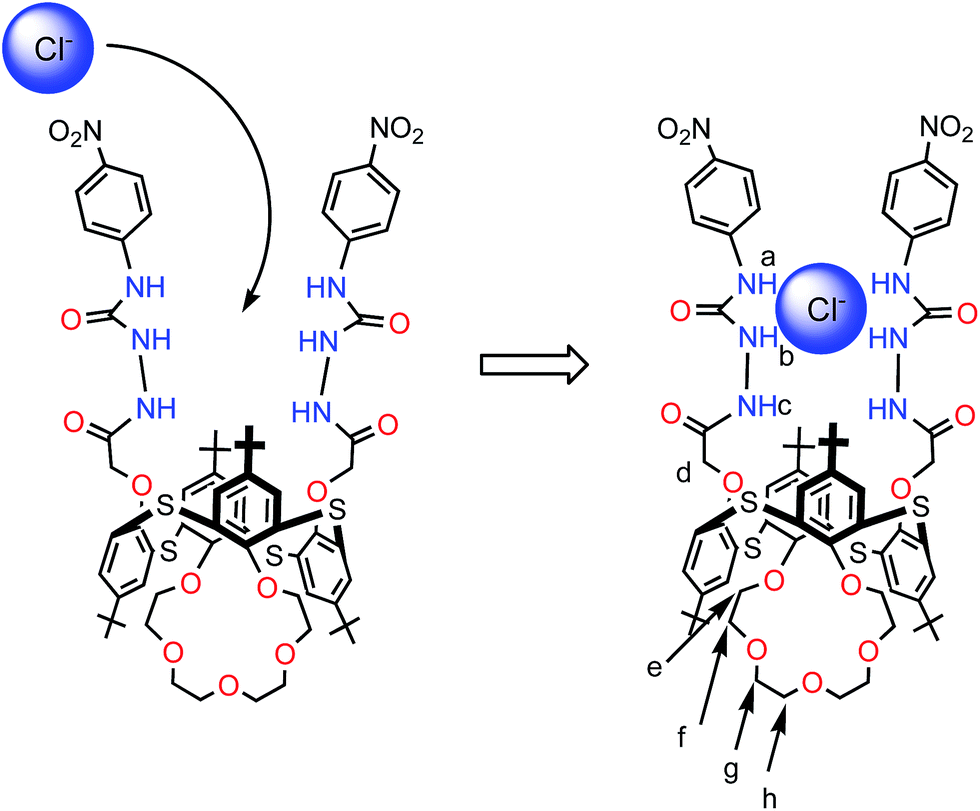
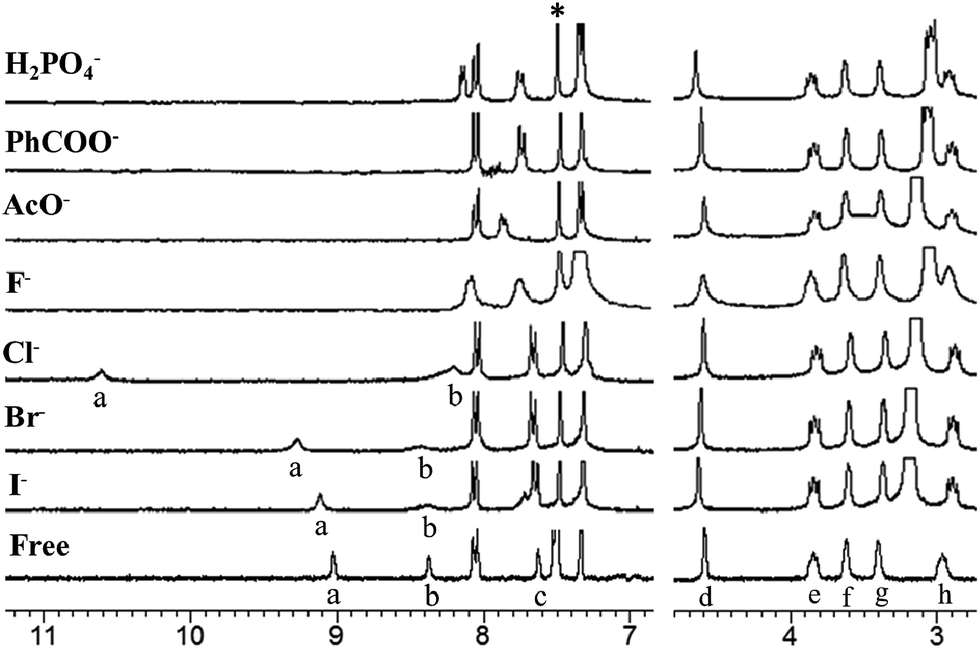
The molecular structures of receptors 4b and 4e were also verified by X-ray crystallographic analysis (Fig. 1 and S15 and S16†). Receptors 4b and 4e were recrystallized from a mixture of CHCl3–CH3CN (1:1, v/v) by slow evaporation. These results indicate that receptors 4b and 4e adopt the 1,3-alternate conformation in the solid state. There are two thiacalixarenes, one water molecule and three chloroform molecules in the asymmetric unit. Interestingly, it was found that two urea groups approach each other and are oriented in parallel due to the existence of dual intramolecular hydrogen bonding (in case of receptor 4e, for the molecule shown: N(14)–H(14)⋯O(21) 2.37(2); N(15)–H(15)⋯O(21) 2.05(2) Å; for the second molecule: N(2)–H(2)⋯O(10) 2.37, N(3)–H(3)⋯O(10) 1.94(2) Å) (Fig. 1 and S16†). Moreover, the thiacalix[4]arene-monocrown-5 has a three-dimentional cavity and is large enough to accommodate the metal cation. The association constants (Ka values) between the receptors 4a–e and Cl− ion were determined by 1H NMR spectroscopic titration experiments (Table 1). These results suggest that the association constants depend on the electron-donating/withdrawing groups located at the m-, or p-positions. In the presence of the electron-withdrawing groups, such as CF3 (receptors 4c and 4d) and NO2 (receptor 4e), the Ka values were greater than that for the unsubstituted receptor (receptor 4a). In contrast, in the case of receptor 4b, possessing the electron-donating Me group, there was a general decrease in the Ka value upon complexation with Cl− ion in comparison with the unsubstituted receptor 4a. Therefore, the introduction of electron-withdrawing groups at the m-, or p-positions appears to increase the acidity of the urea protons, and hence enhance the anion-binding ability through hydrogen-bonding interactions. The Ka value of receptor 4e with the electron-withdrawing NO2 group at the p-position was the best out of all the Ka values measured for receptors 4a–e and Cl− ion. Interestingly, it was found that the Ka value of receptor 4c with the electron-withdrawing CF3 group at the p-position was greater than that of receptor 4d with the electron-withdrawing CF3 group at the m-position. This result indicates that electron-withdrawing groups located at the p-position can significantly influence the acidity of the urea protons by conjugating with the phenyl groups. From the above, it is clear that receptor 4e with the electron-withdrawing NO2 group at the p-position has the most effective recognition ability toward selected anions. Given this, further complexation studies of receptor 4e (2.5 μM) exhibits an absorption band at 310 nm in the UV spectrum in the absence of anions. Upon addition of Cl− ion (0–50 μM) to the solution of receptor 4e, Fig. 2 reveals a gradual decrease in the absorption of the band at 310 nm with a simultaneous increase in the absorption at 340 nm. Meanwhile, a clear isosbestic point was observed at 322 nm for the receptor 4e. A Job's plot binding between the receptor 4e and Cl− ion reveals a 1:1 stoichiometry (Fig. S25†), whilst the association constant (Ka value) for the complexation with Cl− ion by receptor 4e was determined to be 34152 M−1 by UV-vis titration experiments in CHCl3–DMSO (10:1, v/v) (Fig. S24, S27–S31†). Moreover, the concentration dependence of the 1H NMR chemical shifts of the ureido protons in receptor 4e was not observed (Fig. S23†). This result suggests that receptor 4e has a strong intramolecular hydrogen bond between the two ureas linking the p-nitrophenyl moieties. These results strongly suggested that Cl− ion recognition by receptor 4e was via a hydrogen-bonding interaction between the Cl− ion and N–H protons as shown in Fig. 3. Similarly, the UV-vis titration experiments of receptor 4e with other various anions besides Cl− ion were carried out, and the Ka values are summarized in Table 2. As a result, it was found that receptor 4e exhibited high selectivity towards F− ion amongst all of the anions tested, and was capable of complexing with all of the anions tested, irrespective of their shape. Interestingly, the color of the receptor 4e solution changed from colorless to dark yellow upon addition of F− ion (5 equivalents), and this could be easily observed by the naked eye. Upon the addition of F− ions (0–50 μM) to the solution of the receptor 4e, the absorption peak at 342 nm gradually moved to a longer wavelength, finally reaching a maximum value at 360 nm (Fig. 4 and S26†). This result suggests that the quinoid structure was formed by the deprotonation of urea NH groups in the p-nitrophenyl ureido moiety. Moreover, the addition of F−, AcO−, PhCOO− or H2PO4− (1 equivalent) to solutions of receptor 4e in CHCl3–DMSO (10:1, v/v) during the 1H NMR titration experiments resulted in the disappearance of the urea proton signals, NHa and NHb (Fig. 5). These results indicate that strong interactions between these anions and the urea NH groups in the receptor 4e occur and that the kinetics of these anion exchanges is on the NMR time scale. On the other hand, 1H NMR spectroscopic and UV-vis titration experiments of receptor 4e with K+ ion at the crown-ether moiety were also carried out (Fig. S32 and S33†). When only K+ ion (1 equivalent) were added, not only the downfield shift of the crown-ether bridge protons was observed, but also all the NH protons in 1H NMR titration experiments (Fig. 6b and 7b). It was found that a Job's plot binding between receptor 4e and K+ ion exhibited a 1:1 stoichiometry and that the Ka value for the complexation with K+ ion was determined to be 28536 (±1998) M−1 by UV-vis titration experiments in CH2Cl2–DMSO (10:1, v/v) (Fig. S34 and S35†). These results suggest that the crown-5 ring of receptor 4e binds K+ ion. To seek more detailed information about the presence of an effective positive or negative allosteric effect between receptor 4e·K+ and Br− or Cl− ions, 1H NMR spectroscopic and UV-vis titration experiments in CHCl3–DMSO (10:1, v/v) (Fig. S36†) were carried. Fig. 6 reveals that when Br− ion were added to the solution of [4e⊃KSO3CF3] (Fig. 6c), the addition induces a downfield shift of 0.42 ppm (δ = 9.09 to 9.51 ppm) for the NHa protons, and upfield shifts of 0.85 ppm (δ = 8.95 to 8.10 ppm) for the NHb protons and of 0.29 ppm (δ = 8.10 to 7.81 ppm) for the NHc protons, while the chemical shifts for the crown-ether bridge protons did not change. These results suggested the formation of a heteroditopic dinuclear complex of the type Br−⊂[4e⊃K+] (Fig. 6c), and we propose a positive allosteric effect of receptor 4e towards Br− ions in the presence of K+ ion by an ion-pair electrostatic interaction and a conformational change of the flexible thiacalix[4]arene cavity as shown in Fig. 6. On the other hand, Fig. 7 shows that when Cl− ions were added to the solution of [4e⊃KSO3CF3] (Fig. 7c), this addition induces a downfield shift of 1.11 ppm (δ = 9.09 to 10.2 ppm) for the NHa protons and 0.04 ppm (δ = 8.10 to 8.14 ppm) for the NHc protons, and an upfield shift of 0.37 ppm (δ = 8.95 to 8.58 ppm) for the NHb protons, together with upfield shifts for the crown-ether bridge protons. Interestingly, when Cl− ions were added to the solution of [4e⊃KSO3CF3] (Fig. 7c), the chemical shifts for the crown-ether bridge protons most closely matched the chemical shifts for the free crown-ether bridge protons (Fig. 7c and d). These results suggested that the two urea groups in two p-nitrophenyl ureido moieties of receptor 4e·K+ bind the Cl− ion by an ion-pair electrostatic interaction and a conformational change of the flexible thiacalix[4]arene cavity. This induces the decomplexation of the K+ ion from the crown-5 ring of receptor 4e because the Cl− ion has a smaller ionic radius and therefore an increase in basicity in comparison with the Br− ion, and a negative allosteric effect of receptor 4e to Cl− ion in the presence of K+ ion as shown in Fig. 7 is proposed.
 |
| | Fig. 1 X-ray crystal structure of receptor 4e. H-bonds shown as dashed lines. One of two similar molecules in the asymmetric unit is shown in two orientations rotated by approx. 90°. H atoms not involved in H-bonding, minor disorder components, and chloroform molecules of crystallization are omitted for clarity. | |
Table 1 Association constants of receptor 4a–e with Cl− ionsa,b
|
Measured in CDCl3–DMSO (10:1, v/v) at 298 K by the 1H NMR titration method using the chemical-shift change of the NHa proton (Fig. S17–S22); host concentration was 4.0 × 10−3 M.
Guests used: Bu4NCl.
|
| Host |
4a
|
4b
|
4c
|
4d
|
4e
|
| R |
H |
p-CH3 |
p-CF3 |
3,5-(CF3)2 |
p-NO2 |
|
K
a [M−1] |
6816 ± 545 |
3021 ± 242 |
12813 ± 1025 |
6945 ± 625 |
34411 ± 2400 |
 |
| | Fig. 2 UV-vis absorption spectra of receptor 4e (2.5 μM) upon the addition of Bu4NCl (0–50 μM) in CH2Cl2–DMSO (10:1, v/v). | |
 |
| | Fig. 3 Binding mode of receptor 4e upon complexation with Cl− ions. | |
Table 2 Association constants of receptor 4e with various anionsa,b
|
Measured in CH2Cl2–DMSO (10:1, v/v) at 298 K by UV-vis titration method (Fig. 2, 4, S24 and S27–S31); host concentration was 2.5 μM.
Guests used: tetrabutylammonium salt.
|
| Anion |
F− |
Cl− |
Br− |
I− |
AcO− |
PhCO2− |
H2PO4− |
| Shape |
Spherical |
Spherical |
Spherical |
Spherical |
Y-shape |
Y-shape |
Tetrahedral |
|
K
a [M−1] |
128775 ± 10302 |
34152 ± 2732 |
7296 ± 584 |
4540 ± 363 |
107298 ± 8584 |
106743 ± 8539 |
108687 ± 8695 |
 |
| | Fig. 4 UV-vis absorption spectra of receptor 4e (2.5 μM) upon the addition of Bu4NF (0–50 μM) in CH2Cl2–DMSO (10:1, v/v). | |
 |
| | Fig. 5 Partial 1H NMR spectra of receptor 4e/guest (H/G = 1:1); free receptor 4e and in the presence of 1 equiv. of Bu4NX (X = F, Cl, Br, I, AcO, PhCOO, H2PO4). Host concentration was 2.5 μM. Solvent: CDCl3–DMSO (10:1, v/v). 300 MHz at 298 K. *Denotes the solvent peak. | |
 |
| | Fig. 6 Proposed positive allosteric behaviour of receptor 4e with Br− and K+ ions. Partial 1H NMR spectra of 4e/guest (H/G = 1:1); (a) free 4e; (b) 4e⊃KSO3CF3; (c) Bu4NBr⊂[4e⊃K+]; (d) 4e⊃Bu4NBr. Solvent: CDCl3–DMSO (10:1, v/v). 300 MHz at 298 K. *Denotes the solvent peak. | |
 |
| | Fig. 7 Proposed negative allosteric behaviour of 4e with Cl− and K+ ions. Partial 1H NMR spectra of 4e/guest (H/G = 1:1); (a) free 4e; (b) 4e⊃KSO3CF3; (c) Bu4NCl⊂[4e⊃K+]; (d) 4e⊃Bu4NCl. Solvent: CDCl3–DMSO (10:1, v/v). 300 MHz at 298 K. *Denotes the solvent peak. | |
Conclusion
In summary, a new family of heteroditopic receptors (4a–e) based on a thiacalix[4]arene in the 1,3-alternate conformation, which has two ureas moieties bearing various phenyl groups substituted with either electron-donating or -withdrawing groups at their m-, or p-positions, as well as a crown-ether moiety at the opposite side of thiacalix[4]arene cavity, has been synthesized. By using 1H NMR spectroscopic and UV-vis titration experiments, receptor 4e possessing an electron-withdrawing NO2 group at the p-position has the most effective recognition ability towards the selected anions. The binding of K+ ions and various anions at the crown-5 ring moiety and the two urea NH groups in two p-nitrophenyl ureido moieties, respectively, was investigated. The results indicated the complexation mode, and it was found that receptor 4e was able to bind all of the anions tested, irrespective of their shape. Receptor 4e exhibited highest selectivity towards F− ion amongst all of the anions tested and indicated that this receptor might be a promising candidate as a colorimetric chemosensor. The appearance of positive and negative allosteric effects in receptor 4e was also investigated by 1H NMR and UV-vis titration experiments. Interestingly, the formation of a heteroditopic dinuclear complex of receptor 4e with Br− and K+ ions by a positive allosteric effect could be observed. On the other hand, the fact that two urea NH groups in two p-nitrophenyl ureido moieties of receptor 4e·K+ bind the Cl− ion, which then induces the decomplexation of the K+ ion from the crown-5 ring, is indicative of a negative allosteric effect.
Experimental section
General
All melting points were determined with Yanagimoto MP-S1. 1H-NMR spectra were determined at 300 MHz with a Nippon Denshi JEOL FT-300 NMR spectrometer with SiMe4 as an internal reference; J-values are given in Hz. UV spectra were measured by a Shimadzu 240 spectrophotometer. Mass spectra were obtained on a Nippon Denshi JMS-01SG-2 mass spectrometer at an ionization energy of 70 eV using a direct inlet system through GLC. Elemental analyses were performed by Yanaco MT-5.
Materials
Unless otherwise stated, all other reagents used were purchased from commercial sources and used without further purification. Compounds 113 and 212 were prepared following the reported procedures.
Synthesis of compound 3
Compound 2 (1.0 g, 0.95 mmol) was put into a round-bottom flask and ethanol (120 mL), THF (120 mL) and hydrazine hydrate (14 mL, large excess) were added and refluxed for 48 h. After cooling, the solvents and excess hydrazine were removed under reduced pressure to give the crude product as a white solid. The residue was triturated sequentially with water and methanol and the product collected by filtration. Compound 3 was obtained 0.84 g (86%) as a white solid. M.p. 216–218 °C. IR: νmax (KBr)/cm−1: 3421, 2961, 1670, 1438, 1263, 1091, 1019 and 801. 1H NMR (300 MHz, CDCl3): δ = 1.25 (18H, s, tBu × 2), 1.37 (18H, s, tBu × 2), 3.00 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.39 (4H, br, OCH2 × 2), 3.48 (4H, br, NH2 × 2), 3.60 (4H, broad s, OCH2 × 2), 3.96 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.55 (4H, s, OCH2CO × 2), 7.35 (4H, s, Ar-H × 2), 7.41 (4H, s, Ar-H × 2) and 7.54 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3): δ = 30.5 (CH3), 33.5 (C(CH3)3), 64.9 (OCH2), 67.4 (OCH2), 69.2 (OCH2), 70.5 (OCH2), 72.6 (OCH2), 126.2 (ArC), 126.4 (ArC), 126.5 (ArC), 126.7 (ArC), 146.5 (ArC), 146.7 (ArC), 153.6 (ArC), 155.4 (ArC) and 167.6 (CO) ppm. FABMS: m/z: 1023.38 (M+). C52H70N4O9S4 (1023.39): calcd C 61.03, H 6.89, N 5.47. Found: C 61.33, H 6.79, N 5.57.
Synthesis of receptor 4a
To compound 3 (150 mg, 0.147 mmol) in THF (10 mL), was added phenyl isocyanate (38 mg, 0.320 mmol) and the mixture was stirred for at room temperature for 24 h under argon. The resulting precipitate was collected by filtration, washed with hexane to give receptor 4a as a white solid. Recrystallization from CHCl3–CH3CN (4:1) gave receptor 4a (154 mg, 83%) as white solid. M.p. 202–205 °C. IR: νmax (KBr)/cm−1: 3270, 2956, 1674, 1547, 1442, 1263, 1221, 1153, 1091, 799 and 751. 1H NMR (300 MHz, CDCl3–DMSO, 10:1): δ = 1.25 (18H, s, tBu × 2), 1.39 (18H, s, tBu × 2), 2.97 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, s, OCH2 × 2), 3.85 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.59 (4H, s, OCH2CO × 2), 6.95 (2H, t, J = 7.3 Hz, phenyl-H × 2), 7.15 (4H, t, J = 7.6 Hz, phenyl-H × 4), 7.31 (4H, d, J = 7.7 Hz, phenyl-H × 2), 7.35 (4H, s, Ar-H × 4), 7.48 (4H, s, Ar-H × 4), 7.57 (2H, s, NH × 2), 8.10 (2H, s, NH × 2), 8.32 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3–DMSO, 10:1): δ = 29.9 (CH3), 30.4 (CH3), 33.5 (C(CH3)3), 33.5 (C(CH3)3), 64.9 (OCH2), 67.5 (OCH2), 69.0 (OCH2), 70.6 (OCH2), 72.6 (OCH2), 118.4 (ArC), 120.4 (ArC), 121.8 (ArC), 125.6 (ArC), 126.1 (ArC), 126.3 (ArC), 127.1 (ArC), 127.5 (ArC), 128.0 (ArC), 128.2 (ArC), 137.3 (ArC), 146.4 (ArC), 147.5 (ArC), 153.7 (ArC), 154.0 (CO), 154.6 (ArC) and 167.5 (CO) ppm. FABMS: m/z: 1261.43 (M+). C66H80N6O11S4 (1260.48): calcd C 62.83, H 6.39, N 6.66. Found: C 62.59, H 6.23, N 6.45.
Synthesis of receptor 4b
To compound 3 (150 mg, 0.147 mmol) in THF (10 mL), was added p-tolyl isocyanate (43 mg, 0.320 mmol) and the mixture was stirred for at room temperature for 24 h under argon. The resulting precipitate was collected by filtration, washed with EtOH to give receptor 4b as a white solid. Recrystallization from CHCl3–CH3CN (2:1) gave receptor 4b (163 mg, 86%) as white solid. M.p. 205–207 °C. IR: νmax (KBr)/cm−1: 3283, 2955, 1678, 1547, 1444, 1266, 1207, 1151, 1089, 999 and 815. 1H NMR (300 MHz, CDCl3–DMSO, 10:1): δ = 1.27 (18H, s, tBu × 2), 1.39 (18H, s, tBu × 2), 2.28 (6H, s, CH3 × 2), 2.97 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, s, OCH2 × 2), 3.85 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.58 (4H, s, OCH2CO × 2), 6.96 (4H, d, J = 7.7 Hz, phenyl-H × 4), 7.16 (4H, d, J = 7.7 Hz, phenyl-H × 4), 7.35 (4H, s, Ar-H × 4), 7.48 (4H, s, Ar-H × 4), 7.51 (2H, s, NH × 2), 8.10 (2H, s, NH × 2), 8.22 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3–DMSO, 10:1): δ = 20.7 (CH3), 30.9 (CH3), 31.4 (CH3), 34.4 (C(CH3)3), 34.5 (C(CH3)3), 65.9 (OCH2), 68.6 (OCH2), 70.0 (OCH2), 71.6 (OCH2), 73.6 (OCH2), 119.4 (ArC), 126.6 (ArC), 127.0 (ArC), 127.3 (ArC), 128.2 (ArC), 129.0 (ArC), 129.5 (ArC), 131.9 (ArC), 135.7 (ArC), 136.1 (ArC), 147.4 (ArC), 148.5 (ArC), 154.4 (ArC), 154.8 (ArC), 155.1 (CO), 155.5 (ArC) and 168.5 (CO) ppm. FABMS: m/z: 1289.46 (M+). C68H84N6O11S4 (1289.69): calcd C 63.33, H 6.56, N 6.52. Found: C 62.56, H 6.56, N 6.25.
Synthesis of receptor 4c
To compound 3 (150 mg, 0.147 mmol) in THF (10 mL), was added p-trifluoromethylphenyl isocyanate (59 mg, 0.320 mmol) and the mixture was stirred for at room temperature for 24 h under argon. The resulting precipitate was collected by filtration, washed with EtOH to give receptor 4c as a white solid. Recrystallization from CHCl3–CH3CN (1:1) gave receptor 4c (164 mg, 80%) as white solid. M.p. 207–210 °C. IR: νmax (KBr)/cm−1: 3283, 2959, 1687, 1548, 1445, 1266, 1158, 1091, 1068 and 840. 1H NMR (300 MHz, CDCl3–DMSO, 10:1): δ = 1.27 (18H, s, tBu × 2), 1.40 (18H, s, tBu × 2), 2.97 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, s, OCH2 × 2), 3.85 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.61 (4H, s, OCH2CO × 2), 7.36 (4H, s, Ar-H × 4), 7.39–7.42 (8H, m, phenyl-H × 8), 7.49 (4H, s, Ar-H × 4), 7.56 (2H, s, NH × 2), 8.29 (2H, s, NH × 2), 8.69 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3–DMSO, 10:1): δ = 30.9 (CH3), 31.3 (CH3), 34.4 (C(CH3)3), 34.5 (C(CH3)3), 66.1 (OCH2), 68.5 (OCH2), 69.9 (OCH2), 71.6 (OCH2), 73.5 (OCH2), 118.1 (ArC), 122.9 (ArC), 123.9 (CF3), 124.2 (CF3), 125.6 (ArC), 125.8 (ArC), 125.9 (ArC), 126.4 (ArC), 126.9 (ArC), 127.0 (ArC), 128.2 (ArC), 147.4 (ArC), 148.4 (ArC), 154.6 (ArC), 154.8 (CO), 155.5 (ArC) and 167.5 (CO) ppm. FABMS: m/z: 1397.44 (M+). C68H78F6N6O11S4 (1397.63): calcd C 58.44, H 5.63, N 6.01. Found: C 58.62, H 5.53, N 6.13.
Synthesis of receptor 4d
To compound 3 (150 mg, 0.147 mmol) in THF (10 mL), was added 3,5-bis(trifluoromethyl)phenyl isocyanate (82 mg, 0.320 mmol) and the mixture was stirred for at room temperature for 24 h under argon. The resulting precipitate was collected by filtration, washed with EtOH to give receptor 4d as a white solid. Recrystallization from CHCl3–CH3CN (1:1) gave receptor 4d (187 mg, 83%) as white solid. M.p. 208–210 °C. IR: νmax (KBr)/cm−1: 3315, 2963, 1677, 1577, 1443, 1215, 1136, 1092, 1019 and 880. 1H NMR (300 MHz, CDCl3–DMSO, 10:1): δ = 1.32 (18H, s, tBu × 2), 1.39 (18H, s, tBu × 2), 3.01 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.64 (4H, s, OCH2 × 2), 3.89 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.63 (4H, s, OCH2CO × 2), 7.28 (2H, s, phenyl-H × 2), 7.38 (4H, s, Ar-H × 4), 7.42 (4H, s, phenyl-H × 4), 7.49 (4H, s, Ar-H × 4), 7.82 (2H, s, NH × 2), 8.49 (2H, s, NH × 2), 9.05 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3–DMSO, 10:1): δ = 30.8 (CH3), 31.2 (CH3), 34.3 (C(CH3)3), 34.4 (C(CH3)3), 65.8 (OCH2), 67.9 (OCH2), 69.7 (OCH2), 71.4 (OCH2), 73.4 (OCH2), 115.3 (ArC), 117.7 (ArC), 121.6 (ArC), 124.3 (CF3), 126.1 (ArC), 126.7 (ArC), 127.0 (ArC), 127.9 (ArC), 131.7 (ArC), 140.4 (ArC), 147.4 (ArC), 148.4 (ArC), 154.1 (ArC), 154.5 (ArC), 155.4 (CO), 155.5 (ArC) and 167.4 (CO) ppm. FABMS: m/z: 1533.48 (M+). C70H76F12N6O11S4 (1533.63): calcd C 54.82, H 4.99, N 5.48. Found: C 54.63, H 5.05, N 5.35.
Synthesis of receptor 4e
To compound 3 (150 mg, 0.147 mmol) in THF (10 mL), was added p-nitrophenyl isocyanate (53 mg, 0.320 mmol) and the mixture was stirred for at room temperature for 24 h under argon. The resulting precipitate was collected by filtration, washed with EtOH to give receptor 4e as a pale yellow solid. Recrystallization from CHCl3–CH3CN (3:1) gave receptor 4e (165 mg, 83%) as pale yellow solid. M.p. 212–215 °C. IR: νmax (KBr)/cm−1: 3257, 2957, 1682, 1555, 1512, 1445, 1415, 1266, 1150, 1091 and 850. 1H NMR (300 MHz, CDCl3–DMSO, 10:1): δ = 1.27 (18H, s, tBu × 2), 1.39 (18H, s, tBu × 2), 2.97 (4H, t, J = 9.1 Hz, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, s, OCH2 × 2), 3.85 (4H, t, J = 9.1 Hz, OCH2 × 2), 4.58 (4H, s, OCH2CO × 2) 7.40 (4H, s, Ar-H × 4), 8.57 (4H, s, Ar-H × 4), 7.58 (4H, d, J = 9.3 Hz, phenyl-H × 4), 7.66 (2H, s, NH × 2), 8.06 (4H, d, J = 9.3 Hz, phenyl-H × 4), 8.40 (2H, s, NH × 2), 9.08 (2H, s, NH × 2) ppm. 13C NMR (100 MHz, CDCl3–DMSO, 10:1): δ = 30.9 (CH3), 31.3 (CH3), 34.4 (C(CH3)3), 34.5 (C(CH3)3), 66.2 (OCH2), 69.0 (OCH2), 69.9 (OCH2), 71.7 (OCH2), 73.6 (OCH2), 118.1 (ArC), 124.9 (ArC), 126.2 (ArC), 127.1 (ArC), 127.7 (ArC), 128.0 (ArC), 128.4 (ArC), 142.6 (ArC), 144.5 (ArC), 147.5 (ArC), 147.3 (ArC), 147.9 (ArC), 148.2 (ArC), 154.0 (ArC), 154.2 (CO), 155.7 (ArC) and 168.5 (CO) ppm. FABMS: m/z: 1351.57 (M+). C66H78N8O15S4 (1351.63): calcd C 58.65, H 5.82, N 8.29. Found: C 58.81, H 5.75, N 8.12.
Determination of the association constants
The association constants were determined by using 1H NMR spectroscopic titration experiments in a constant concentration of host receptor (4.0 × 10−3 M) and varying the guest concentration (0–8.0 × 10−3 M). The 1H NMR chemical shift of the urea protons (NH) signal was used as a probe. The association constant (Ka) for the complexes of receptor 4a–e were calculated by nonlinear curve-fitting analysis of the observed chemical shifts of the NH protons according to the literature procedure.14
1H NMR titration experiments
A solution of Bu4NX (X = F, Cl, Br, I, AcO, PhCOO, H2PO4) in CD3CN (4.0 × 10−3 M) was added to a CDCl3 solution of receptor 4a–e in the absence or presence of KSO3CF3 in an NMR tube. 1H NMR spectra were recorded after addition of the reactants and the temperature of the NMR probe was kept constant at 27 °C. The 1H NMR spectroscopic data of representative complexes are given below:
Receptor 4a⊃Cl−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.59 (4H, s, OCH2O × 2), 7.89 (2H, br, NHc × 2), 8.10 (2H, br, NHb × 2) and 8.95 (2H, br, NHa × 2) ppm.
Receptor 4b⊃Cl−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.68 (4H, s, OCH2O × 2), 7.80 (2H, br, NHc × 2), 8.09 (2H, br, NHb × 2) and 8.63 (2H, br, NHa × 2) ppm.
Receptor 4c⊃Cl−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.68 (4H, s, OCH2O × 2), 8.01 (2H, br, NHc × 2), 8.20 (2H, br, NHb × 2) and 9.58 (2H, br, NHa × 2) ppm.
Receptor 4d⊃Cl−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 3.01 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.64 (4H, br, OCH2 × 2), 3.89 (4H, br, OCH2 × 2), 4.63 (4H, s, OCH2O × 2), 7.94 (2H, br, NHc × 2), 8.33 (2H, br, NHb × 2) and 9.70 (2H, br, NHa × 2) ppm.
Receptor 4e⊃Cl−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.60 (4H, s, OCH2O × 2), 8.10 (2H, br, NHc × 2), 8.18 (2H, br, NHb × 2) and 10.8 (2H, br, NHa × 2) ppm.
Receptor 4e⊃K+.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 3.11 (4H, br, OCH2 × 2), 3.36–3.58 (4H, m, OCH2 × 2), 3.64–3.90 (4H, m, OCH2 × 2), 4.08 (4H, br, OCH2 × 2), 4.30–4.61 (4H, m, OCH2O × 2), 8.10 (2H, s, NHc × 2), 8.95 (2H, broad s, NHb × 2) and 9.09 (2H, broad s, NHa × 2) ppm.
Cl−⊂[receptor 4e⊃K+].
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.60 (4H, s, OCH2O × 2), 8.14 (2H, br, NHc × 2), 8.58 (2H, br, NHb × 2) and 10.2 (2H, br, NHa × 2) ppm.
Receptor 4e⊃Br−.
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 2.97 (4H, br, OCH2 × 2), 3.40 (4H, br, OCH2 × 2), 3.63 (4H, br, OCH2 × 2), 3.85 (4H, br, OCH2 × 2), 4.60 (4H, s, OCH2O × 2), 7.52 (2H, br, NHc × 2), 8.25 (2H, br, NHb × 2) and 9.27 (2H, br, NHa × 2).
Br−⊂[receptor 4e⊃K+].
1H NMR (300 MHz, CHCl3–DMSO–CH3CN, 10:1:1, v/v): δ = 3.11 (4H, br, OCH2 × 2), 3.36–3.58 (4H, m, OCH2 × 2), 3.64–3.90 (4H, m, OCH2 × 2), 4.08 (4H, br, OCH2 × 2), 4.30–4.61 (4H, m, OCH2O × 2), 7.81 (2H, br, NHc × 2), 8.10 (2H, br, NHb × 2) and 9.51 (2H, br, NHa × 2).
Crystallographic analysis of receptors 4b and 4e
Crystal data for 4b.
C68H84N6O11S4·½(H2O)·1½(CHCl3), Mr = 1477.71. Monoclinic, P21/n; a = 18.8935 (13), b = 23.9302 (16), c = 33.589 (2) Å; β = 91.5063 (12)°; V = 15181.2 (17) Å3; Z = 8; Dx = 1.293 Mg m−3; F(000) = 6224; T = 210(2) K; μ (Mo-Kα) = 0.34 mm−1; λ = 0.71073 Å, crystal size 0.71 × 0.54 × 0.32 mm3. Crystals were colorless blocks. Diffraction data were measured on a Bruker APEX 2 CCD diffractometer equipped with graphite monochromated MoKα radiation by thin-slice ω-scans.15 134900 measured reflections, 31218 independent reflections (Rint = 0.049) to θmax = 26.5°; 19539 reflections with I > 2σ(I). The structure was determined by direct methods using the SHELXS program and refined by the full-matrix least-squares method, on F2, in SHELXL-2013/14.16,17 The non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms on C were included in idealized positions and their Uiso values were set to ride on the Ueq values of the parent atoms. H atoms on N were freely refined. At the conclusion of the refinement, wR2 = 0.173 (all data) and R1 = 0.056 (observed data), 1903 parameters, Δ〉max = 0.56 eÅ−3; 465 restraints, Δ〉min = −0.43 eÅ−3. The platon squeeze procedure was used to model two of the three unique CHCl3 molecules due to severe disorder.18 Two-fold disorder was modelled in some tBu groups, in parts of one of the crown ether chains and the other CHCl3 molecule. H atoms on water molecule O(23) could not be located in difference maps, so were not included in the model.†
Crystal data for 4e.
C66H78N8O15S4·½(CHCl3)·3(MeCN), Mr = 1534.44. Monoclinic, P21/c; a = 17.7980 (10), b = 26.7870 (16), c = 32.552 (2) Å; β = 96.384 (4)°; V = 15423.1 (16) Å3; Z = 8; Dx = 1.322 Mg m−3; F(000) = 6472; T = 100 (2) K; μ (Mo-Kα) = 0.31 mm−1; λ = 0.7749 Å, crystal size 0.25 × 0.25 × 0.02 mm3. Crystals were colorless plates. Diffraction data were measured on a Bruker APEX 2 CCD diffractometer at station 11.3.1 of the ALS using synchrotron radiation by thin-slice ω-scans.15 155885 measured reflections, 50956 independent reflections (Rint = 0.052) to θmax = 34.8°; 35702 reflections with I > 2σ(I). Structure solution with SHELXT and refinement as above.16,17 Hydrogen atoms on C and some N atoms were included in idealized positions and their Uiso values were set to ride on the Ueq values of the parent atoms. H atoms on the remaining N atoms were freely refined. At the conclusion of the refinement, wR2 = 0.294 (all data) and R1 = 0.086 (observed data), 2055 parameters, Δ〉max = 2.44 eÅ−3; 656 restraints, Δ〉min = −1.86 eÅ−3. The platon squeeze procedure was used to model four of the six unique MeCN molecules due to severe disorder.18 Two-fold disorder was modelled in some tBu groups and in parts of one the crown ether chains and one HN-p-C6H4NO2 group.†
Acknowledgements
This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices (Institute for Materials Chemistry and Engineering, Kyushu University)”. We would like to thank the OTEC at Saga University and the International Cooperation Projects of Guizhou Province (no. 20137005) for financial support. CR thanks the EPSRC for a travel grant. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231.
Notes and references
-
(a)
C. D. Gutsche, Calixarenes, An Introduction, Royal Society of Chemistry, Cambridge, UK, 2008 Search PubMed;
(b) A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS PubMed;
(c) D. Coquière, S. Le Gac, U. Darbost, O. Sénèque, I. Jabin and O. Reinaud, Org. Biomol. Chem., 2009, 7, 2485–2500 RSC;
(d) K. Cottet, P. M. Marcos and P. J. Cragg, Beilstein J. Org. Chem., 2012, 8, 201–226 CrossRef CAS PubMed;
(e) L. Mutihac, J. H. Lee, J. S. Kim and J. Vicens, Chem. Soc. Rev., 2011, 40, 2777–2796 RSC;
(f) L. Baldini, A. Casnati, F. Sansone and R. Ungaro, Chem. Soc. Rev., 2007, 36, 254–266 RSC;
(g) J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed;
(h) R. Joseph and C. P. Rao, Chem. Rev., 2011, 111, 4658–4702 CrossRef CAS PubMed;
(i) C. Capici, Y. Cohen, A. D'Urso, G. Gattuso, A. Notti, A. Pappalardo, S. Pappalardo, M. F. Parisi, R. Purrello, S. Slovak and V. Villari, Angew. Chem., Int. Ed., 2011, 50, 12162–12167 CrossRef;
(j) C. Talotta, C. Gaeta, Z. Qi, C. A. Schalley and P. Neri, Angew. Chem., Int. Ed., 2013, 52, 7437–7441 CrossRef CAS PubMed;
(k) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed.
- H. Kumagi, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971–3972 CrossRef.
-
(a) P. Lhoták, Eur. J. Org. Chem., 2004, 1675–1692 CrossRef;
(b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed.
-
(a) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS;
(b) T. Nabeshima, T. Saiki and S. Kumitomo, Org. Lett., 2002, 4, 3207–3209 CrossRef CAS PubMed;
(c) T. Nabeshima, Y. Yoshihira, T. Saiki, S. Akine and E. Horn, J. Am. Chem. Soc., 2003, 125, 28–29 CrossRef CAS PubMed;
(d) A. Y. Zhukov, T. A. Fink, I. I. Stoikov and I. S. Antipin, Russ. Chem. Bull., 2009, 58, 1007–1014 CrossRef CAS;
(e) K. Mohr, J. Schmitz, R. Schrage, C. Trnkle and U. Holzgrabe, Angew. Chem., Int. Ed., 2013, 52, 508–516 CrossRef CAS PubMed;
(f) R. Nussinov and C.-J. Tsai, Cell, 2013, 153, 293–305 CrossRef CAS PubMed.
-
(a) J.-Y. Kwon, Y.-J. Jang, S.-K. Kim, K.-H. Lee, J.-S. Kim and J. Yoon, J. Org. Chem., 2004, 69, 5155–5157 CrossRef CAS PubMed;
(b) D. Amilan Jose, D. K. Kumar, B. Ganguly and A. Das, Org. Lett., 2004, 6, 3445–3448 CrossRef PubMed;
(c) J.-Y. Lee, E.-J. Cho, S. Mukamel and K.-C. Nam, J. Org. Chem., 2004, 69, 943–950 CrossRef CAS PubMed;
(d) D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717–5720 CrossRef PubMed;
(e) V. Thiagarajan, P. Ramamurthy, D. Thirumalai and V. T. Ramakrishnan, Org. Lett., 2005, 7, 657–660 CrossRef CAS PubMed;
(f) H. Lu, W. Xu, D. Zhang, C. Chen and D. Zhu, Org. Lett., 2005, 7, 4629–4632 CrossRef CAS PubMed;
(g) F. M. Pfeffer, T. Gunnlaugsson, P. Jensen and P. E. Kruger, Org. Lett., 2005, 7, 5357–5360 CrossRef CAS PubMed;
(h) L. Fang, W.-H. Chan, Y.-B. He, D. W.-J. Kwong and A. W.-M. Lee, J. Org. Chem., 2005, 70, 7640–7646 CrossRef CAS PubMed;
(i) T. Gunnlaugsson, P. E. Kruger, P. Jensen, J. Tierney, H. D. Paduka Ali and G. M. Hussey, J. Org. Chem., 2005, 70, 10875–10878 CrossRef CAS PubMed;
(j) A. Dahan, T. Ashkenazi, V. Kuznetsov, S. Makievski, E. Drug, L. Fadeev, M. Bramson, S. Schokoroy, E. Rozenshine-Kemelmakher and M. Gozin, J. Org. Chem., 2007, 72, 2289–2296 CrossRef CAS PubMed;
(k) S. Saha, A. Ghosh, P. Mahato, S. Mishra, S. K. Mishra, E. Suresh, S. Das and A. Das, Org. Lett., 2010, 12, 3406–3409 CrossRef CAS PubMed.
-
J. L. Sessler, P. A. Gale and W. S. Cho, Anion Receptor Chemistry, Royal Society of Chemistry, Cambridge, U.K., 2006 Search PubMed.
-
(a) J. F. Zhang, Y. Zhou, J. Yoon and J. S. Kim, Chem. Soc. Rev., 2011, 40, 3416–3429 RSC;
(b) C. Lodeiro, J. L. Capelo, J. C. Mejuto, E. Oliveira, H. M. Santos, B. Pedras and C. Nuñez, Chem. Soc. Rev., 2010, 39, 2948–2976 RSC;
(c) L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2013, 42, 3489–3613 RSC.
-
(a) R. M. F. Batista, E. Oliveira, S. P. G. Costa, C. Lodeiro and M. M. M. Raposo, Org. Lett., 2007, 9, 3201–3204 CrossRef CAS PubMed;
(b) F. Zapata, A. Caballero, A. Espinosa, A. Tárraga and P. Molina, Org. Lett., 2008, 10, 41–44 CrossRef CAS PubMed;
(c) R. D. Rasberry, M. D. Smith and K. D. Shimizu, Org. Lett., 2008, 10, 2889–2892 CrossRef CAS PubMed;
(d) C. Pérez-Casas and A. K. Yatsimirsky, J. Org. Chem., 2008, 73, 2275–2284 CrossRef PubMed;
(e) J. P. Clare, A. Statnikov, V. Lynch, A. L. Sargent and J. W. Sibert, J. Org. Chem., 2009, 74, 6637–6646 CrossRef CAS PubMed;
(f) Q.-S. Lu, L. Dong, J. Zhang, J. Li, L. Jiang, Y. Huang, S. Qin, C.-W. Hu and X.-Q. Yu, Org. Lett., 2009, 11, 669–672 CrossRef CAS PubMed;
(g) S. Goswami, D. Sen and N. K. Das, Org. Lett., 2010, 12, 856–859 CrossRef CAS PubMed;
(h) A. Aldrey, C. Nứñez, V. García, R. Bastida, C. Lodeiro and A. Macías, Tetrahedron, 2010, 66, 9223–9230 CrossRef CAS;
(i) P. Dydio, T. Zieliński and J. Jurczak, Org. Lett., 2010, 12, 1076–1078 CrossRef CAS PubMed;
(j) V. K. Bhardwaj, S. Sharma, N. Singh, M. S. Hundal and G. Hundal, Supramol. Chem., 2011, 23, 790–800 CrossRef CAS;
(k) G.-W. Lee, N.-K. Kim and K.-S. Jeong, Org. Lett., 2011, 13, 3024–3027 CrossRef PubMed;
(l) H. M. Chawla, S. N. Sahu, R. Shrivastava and S. Kumar, Tetrahedron Lett., 2012, 53, 2244–2247 CrossRef CAS;
(m) S. Goswami, A. Manna, S. Paul, K. Aich, A. K. Das and S. Chakraborty, Tetrahedron Lett., 2013, 54, 1785–1789 CrossRef CAS;
(n) K. Pandurangan, J. A. Kitchen and T. Gunnlaugsson, Tetrahedron Lett., 2013, 54, 2770–2775 CrossRef CAS;
(o) S. Areti, J. K. Khedkar, R. Chilukula and C. P. Rao, Tetrahedron Lett., 2013, 54, 5629–5634 CrossRef CAS;
(p) C. Jin, M. Zhang, C. Deng, Y. Guan, J. Gong, D. Zhu, Y. Pan, J. Jiang and L. Wang, Tetrahedron Lett., 2013, 54, 796–801 CrossRef CAS.
-
(a) K. Lang, P. Cuřínová, M. Dudič, P. Prošcová, I. Stibor, V. Št'astný and P. Lhoták, Tetrahedron Lett., 2005, 46, 4469–4472 CrossRef CAS;
(b) P. Lhoták, J. Svoboda and I. Stibor, Tetrahedron Lett., 2006, 62, 1253–1257 CrossRef;
(c) J. Kroupa, I. Stibor, M. Pojarová, M. Tkadlecová and P. Lhoták, Tetrahedron, 2008, 64, 10075–10079 CrossRef CAS;
(d) O. Kundrat, H. Dvorakova, I. Cisarova, M. Pojarova and P. Lhoták, Org. Lett., 2009, 11, 4188–4191 CrossRef CAS PubMed;
(e) O. Kundrat, I. Cisarova, S. Böhm, M. Pojarova and P. Lhoták, J. Org. Chem., 2009, 74, 4592–4596 CrossRef CAS PubMed;
(f) O. Kundrat, H. Dvorakova, V. Eigner and P. Lhoták, J. Org. Chem., 2010, 75, 407–411 CrossRef CAS PubMed;
(g) O. Kundrat, J. Kroupa, S. Böhm, J. Budka, V. Eigner and P. Lhoták, J. Org. Chem., 2010, 75, 8372–8375 CrossRef CAS PubMed;
(h) O. Kundrát, V. Eigner, P. Cuřínová, J. Kroupa and P. Lhoták, Tetrahedron, 2011, 67, 8367–8372 CrossRef;
(i) O. Kundrat, V. Eigner, H. Dvorakova and P. Lhoták, Org. Lett., 2011, 13, 4032–4035 CrossRef CAS PubMed;
(j) P. Slavik, M. Dudic, K. Flidrova, J. Sykora, I. Cisarova, M. Pojarova and P. Lhoták, Org. Lett., 2012, 14, 3628–3631 CrossRef CAS PubMed;
(k) O. Kundrat, H. Dvorakova, S. Böhm, V. Eigner and P. Lhoták, J. Org. Chem., 2012, 77, 2272–2278 CrossRef CAS PubMed.
-
(a) V. Bhalla, M. Kumar, H. Katagiri, T. Hattori and S. Miyano, Tetrahedron Lett., 2005, 46, 121–124 CrossRef CAS;
(b) V. Bhalla, J. N. Babu, M. Kumar, T. Hattori and S. Miyano, Tetrahedron Lett., 2007, 48, 1581–1585 CrossRef CAS;
(c) V. Bhalla, R. Kumar, M. Kumar and A. Dhir, Tetrahedron, 2007, 63, 11153–11159 CrossRef CAS;
(d) A. Dhir, V. Bhalla and M. Kumar, Org. Lett., 2008, 10, 4891–4894 CrossRef CAS PubMed;
(e) R. Kumar, V. Bhalla and M. Kumar, Tetrahedron, 2008, 64, 8095–8101 CrossRef CAS;
(f) R. K. Mahajan, R. Kaur, V. Bhalla, M. Kumar, T. Hattori and S. Miyano, Sens. Actuators, B, 2008, 130, 290–294 CrossRef CAS;
(g) J. N. Babu, V. Bhalla, M. Kumar, R. K. Mahajan and R. K. Puri, Tetrahedron Lett., 2008, 49, 2772–2775 CrossRef CAS;
(h) M. Kumar, A. Dhir and V. Bhalla, Org. Lett., 2009, 11, 2567–2570 CrossRef CAS PubMed;
(i) M. Kumar, R. Kumar and V. Bhalla, Tetrahedron, 2009, 65, 4340–4344 CrossRef CAS;
(j) M. Kumar, A. Dhir and V. Bhalla, Org. Lett., 2009, 11, 2567–2570 CrossRef CAS PubMed;
(k) M. Kumar, R. Kumar and V. Bhalla, Tetrahedron Lett., 2010, 51, 5559–5562 CrossRef CAS;
(l) M. Kumar, R. Kumar and V. Bhalla, Org. Lett., 2011, 13, 366–369 CrossRef CAS PubMed;
(m) M. Kumar, R. Kumar and V. Bhalla, Org. Biomol. Chem., 2011, 9, 8237–8245 RSC;
(n) M. Kumar, R. Kumar and V. Bhalla, Org. Lett., 2011, 13, 366–369 CrossRef CAS PubMed;
(o) M. Kumar, R. Kumar and V. Bhalla, Tetrahedron Lett., 2013, 54, 1524–1527 CrossRef CAS.
-
(a) C. Perez-Casas and T. Yamato, J. Inclusion Phenom. Macrocyclic Chem., 2005, 53, 1–8 CrossRef;
(b) T. Yamato, C. Perez-Casas, H. Yamamoto, M. R. J. Elsegood, S. H. Dale and C. Redshaw, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 261–269 CrossRef CAS;
(c) C. Perez-Casas, S. Rahman, N. Begum, Z. Xi and T. Yamato, J. Inclusion Phenom. Macrocyclic Chem., 2008, 60, 173–185 CrossRef CAS;
(d) X.-L. Ni, X. Zeng, C. Redshaw and T. Yamato, J. Org. Chem., 2011, 76, 3358–3370 Search PubMed;
(e) X.-L. Ni, X. Zeng, C. Redshaw and T. Yamato, Tetrahedron, 2011, 67, 3248–3253 CrossRef CAS;
(f) X.-L. Ni, J. Tahara, S. Rahman, X. Zeng, D. L. Hughes, C. Redshaw and T. Yamato, Chem.–Asian J., 2012, 7, 519–527 CrossRef CAS PubMed;
(g) X.-L. Ni, H. Cong, A. Yoshizawa, S. Rahman, H. Tomiyasu, U. Rayhan, X. Zeng and T. Yamato, J. Mol. Struct., 2013, 1046, 110–115 CrossRef CAS.
- F. W. B. van Leewen, H. Beijleveld, H. Kooijman, A. L. Spek, W. Verboom and D. N. Reinhoudt, J. Org. Chem., 2004, 69, 3928–3936 CrossRef PubMed.
- N. Iki, N. Morohashi, F. Narumi, T. Fujimoto, T. Suzuki and S. Miyano, Tetrahedron Lett., 1999, 40, 7337–7341 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
-
APEX 2 and SAINT, Software for CCD diffractometers, Bruker AXS Inc., Madison, USA, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the 1H/13C NMR spectra, 1H NMR spectroscopic and UV-vis titration experimental data, the Bensei–Hilderbrand plot and Job's plot. CCDC 1026081 and 1026090. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15905e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a