
Prediction of quantum anomalous Hall effect on graphene nanomesh
Xiaoming Zhang and
Mingwen Zhao*
School of Physics and State Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, Shandong, China. E-mail: zmw@sdu.edu.cn
First published on 6th January 2015
Abstract
A quantum anomalous Hall effect (QAHE) has been realized in the Cr-doped magnetic topological insulator at an extremely low temperature (∼30 mK). From first-principles, we predict that QAHE can also be achieved in a well-designed graphene nanomesh without the requirement of transition metal doping. The ferromagnetic ordering arises mainly from the in-plane pxy orbitals, leading to a Curie temperature of 830 K. The bulk band gap due to the intrinsic spin–orbital coupling (SOC) of the pxy orbitals is about 3.7 meV, corresponding to an operating temperature of 43 K. This work reveals a viable approach for realizing QAHE in metal-free graphene nanostructures.
Introduction
The quantum anomalous Hall effect (QAHE) is a topological nontrivial phase characterized by a finite Chern number and helical edge states within the bulk band gap. Without an external magnetic field, internal magnetization breaks time-reversal symmetry and spin–orbit coupling (SOC) opens a SOC-gap, giving rise to a quantized anomalous Hall conductivity. The Hall conductivity σxy develops plateaus at values (Chern number) quantized in unit of e2/h. The helical edge states are robust against backscattering and thus promising for low power consumption device applications.Haldane first proposed a honeycomb-net model (known as Haldane model) for achieving QAHE. He demonstrated theoretically that the break of time-reversal invariance does not necessarily require external magnetic fields, but could occur as a consequence of magnetic ordering in a quasi-two-dimensional lattice.1,2 The internal ferromagnetic (FM) ordering splits the spin-degenerated bands of topological insulators (TIs), leading to time-reversal symmetry-breaking. Magnetic topological insulators (TIs) are therefore expected to be promising for achieving QAHE.3–7 In order to induce stable FM ordering, heavy transition metals are included in these systems, such as mercury-based quantum wells,8 topological insulator thin films containing heavy elements,9,10 graphene11–13 and silicene14,15 based systems, and kagome lattice16,17 systems. However, due to the difficulty in controlling magnetization and SOC, the QAHE has not been observed experimentally until very recently, when it was finally realized in a Cr-doped (Bi, Sb)2Te3 topological insulator at extremely low temperature (∼30 mK).18
Meanwhile, increasing attention has been paid to searching for metal-free topological insulators, since the first prediction on the quantum spin Hall (QSH) states of graphene.19 However, the SOC in graphene is extremely weak, leading to a unobservably small topologically nontrivial band gap, which limits the operating regime to unrealistically low temperatures. Several two-dimensional (2D) materials beyond graphene, such as silicene, germanene,20 silicon carbide,21 graphyne,22 carbon nitride,23 have been proposed as candidates for achieving QSH effect at an experimentally accessible temperature. These works enrich the database of 2D TIs and offer additional candidate materials for achieving QAHE by inducing stable FM ordering. Notably, theoretical works have showed that other than transition metal atoms, nitrogen atom can induce stable FM ordering in semiconductors, such as graphitic carbon nitrides24–26 and silicon carbide.27
In this work, based on first-principles calculations, we propose a nitrogen-doped graphene kagome nanomesh as a promising metal-free magnetic topological insulator, which can be used to realize QAHE at high temperature. Combined with a Monte Carlo (MC) simulation and anomalous Hall conductivity calculation, we demonstrate that this nanomesh has stable FM ordering with a Curie temperature of 830 K and a topologically nontrivial band gap of 3.7 meV (∼43 K) characterized by a Chern number of −1. The stable FM and relatively strong SOC are both related to the in-plane pxy atomic orbitals rather than the pz orbital. We also propose a single orbital tight binding (TB) model on an extended ruby lattice (named as extended ruby model) to reproduce the exotic properties of the nanomesh. The high Curie temperature and moderate SOC gap indicate the feasibility of the QAHE in the metal-free magnetic TI at experimentally accessible temperature.
Method and computational details
Our first principles calculations are performed in the framework of density-functional theory (DFT) as implemented in the Vienna ab initio simulation package (VASP).28 The electron–electron interactions are treated within a generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) for the exchange-correlation functional.29 The electron–ion interaction is described by projector-augmented-wave (PAW) potentials. The electron wave functions are expanded using the plane-waves with the energy cutoff of 500 eV. Self-consistent solution of the Kohn–Sham equation is carried out with the convergence of 10−8. The supercells are repeated periodically on the x–y plane while a vacuum region of about 15 Å is applied along the z-direction to avoid mirror interaction between neighboring images. A symmetry reduced 13 × 13 × 1 Monkhorst-Pack sampling is used for the Brillouin zone (BZ) integration. Structural optimizations are performed using a conjugate gradient (CG) method until the remanent force on each atom is less than 0.002 eV Å−1. Electron spin-polarization is involved in all calculations. The phonon spectra are calculated using a finite displacement method, as implemented in the phonon code30,31 combined with the VASP code. The anomalous Hall conductivity was calculated using the Kubo formula based on the maximally localized Wannier functions.32Result and discussion
The optimized nitrogen-doped graphene kagome nanomesh is shown in Fig. 1a. It can be regarded as a porous graphene monolayer with the marginal carbon (C) atoms around the pores being substituted by nitrogen (N) atoms. We chose N dopant here considering that N have been widely used for chemical doping of carbon materials due to a comparable atomic size and five valence electrons for bonding with carbon atoms.33 Meanwhile, N-doping of graphene was highly effective in modulating its band gap to achieve new properties for device applications.34 It is clearly that the planar configuration and six-fold rotational symmetry of the nanomesh are both preserved, displaying characteristics of a structurally perfect kagome lattice. The equilibrium lattice constant is found to be 11.77 Å with an angle of 60° or 120° between the primitive vectors from present calculations. A vibrational analysis of phonon spectrum calculated from first-principles confirms the stability of the hypothetic nanomesh, because no modes with imaginary frequencies are found in the phonon spectrum along the highly symmetric points in BZ as shown in Fig. 1b. | ||
| Fig. 1 (a) Schematic representation and (b) phonon spectrums of the nitrogen-doped graphene kagome nanomesh. | ||
Certainly, the experimental realization of the nanomesh remains challenging for chemists, though it is kinetically stable. Both unified pores in graphene and exactly N-doping are difficult to control in experiments. However, the recent refinements in the techniques of fabricating porous graphene nanomeshs (GNMs)35–38 offer potential pathways to synthesizing a GNM with well-ordered, appropriately-sized pores. Meanwhile, started from the functionalization of graphene with nitrogen achieved by Wang et al.,39 abundant strategies to prepare N-doped graphene has been reported in controlled ways.40–43 The doping nitrogen tend to siting at the edge of pores44,45 provides a promising prospect to controllable synthesis of the nanomesh proposed in Fig. 1a. Notably, the above top-down strategy involves the generation of well-ordered pores in graphene sheets, which may be limited by the quality of GNMs. Bottom-up approaches refer to the construction of nanomesh from small organic precursor molecules through catalytic or thermal treatment. The power of this synthetic approach lies in the fact that it is possible to design molecular precursors leading to uniquely defined structures. This approach was successfully applied to construct a two-dimensional framework from building blocks created via the trimerizaton of terephthalonitrile.46 Hence, the N-doped graphene kagome nanomesh may also be synthesized by the controlled bottom-up approach with appropriate precursor monomers, such as N-doped graphene quantum dots (N-GQDs)47 and melem pre-treated with absolute ethanol.48–50
To understand the electronic properties of nanomesh, we performed electronic band structure calculations. Self-consistent calculations without SOC gave a stable spin-polarized state with a magnetic moment of 2.0 μB in one unit cell (containing a C14N12 formula). The spin-resolved band structures are plotted in Fig. 2a along the highly-symmetric points (K and K′) in BZ. We can clearly see the characteristics of kagome-like bands consisting of one flat band above two Dirac bands. Notably, because of the magnetization, the spin degeneracy of the kagome-like bands is lifted with spin-down channel being well separated from other bands and the Fermi level crossing the Dirac point exactly (a spin-gapless semiconductor). The Dirac points of the two spin channels remain touching at the K or K′ points in BZ, but with a split of about 0.42 eV. The features can be seen more clearly from the orbital-resolved electron density of states (DOS) shown in the right panel of Fig. 2a. Meanwhile, the DOS demonstrates that the local magnetic moments come mainly from the px and py electrons and minor from the s electrons, rather than the pz electrons. This feature is further confirmed by the isosurfaces of Kohn–Sham wave functions of the three spin-polarized kagome-like bands. The shapes of the isosurfaces shown in Fig. 2b display clearly characteristics of hybridized pxy orbitals, which are expected to have stronger SOC effect than pz orbital.23,51
 | ||
| Fig. 2 (a) Spin-resolved band lines (without SOC) and orbital-resolved electron density of states of the nanomesh. (b) The isosurfaces of Kohn–Sham wave functions of the three bands in proximity of the Fermi level with the isosurface value of 0.004 Å−3. | ||
Since the nanomesh is magnetic, it is of interest to study the preferred magnetic coupling between local magnetic moments. We first carefully studied the magnetic interaction in one unit cell and found that there is no stable solution with an antiferromagnetic configuration (AFM), and the ferromagnetic (FM) ordering is the ground state which is more stable than the nonmagnetic (NM) state by about 145.6 meV. This result can be explained from the perspective of the energy levels of the isolated molecules (see Fig. 3a) constituting the nanomesh. After structural relaxation, self-consistent calculations demonstrated that the molecule favors a spin-polarized ground state. For the three energy levels nearest to the Fermi level, the spin-up branch is fully occupied and the spin-down branch is only partially (2/3) occupied, producing a net magnetic moment of 1.0 μB (see Fig. 3a). These partially-occupied states can act as acceptor levels and mediate a virtual hopping between adjacent magnetic moments for the FM arrangement as shown in Fig. 3b, leading to a stable FM state.52
 | ||
| Fig. 3 Schematic diagrams of spin-resolved energy levels exchange mechanisms for (a) isolated molecule and (b) molecular dimer cutting from the nanomesh with the corner carbon passivated by hydrogen. (c) The exchange mechanisms for the graphene nanomesh. (d) FM and AFM orderings of the graphene nanomesh. The direction of magnetic moments are represented by dot or cross in circle. (e) Temperature-dependent average energy and magnetic moments per unit cell. | ||
Consequently, we adopted large-size supercells containing four primitive cells (donated as 2 × 2) to further confirm the magnetic coupling and estimate Curie temperature. Starting from different initial spin arrangements of the local magnetic moments of 2.0 μB, self-consistent calculations lead to two types of magnetic orderings, FM and AFM, as shown in Fig. 3d, respectively. The AFM states can be seen as an antiferromagnetic combination of collinear ferromagnetic configuration. We found that the AFM phase is less stable than the FM phase by about 426.3 meV per 2 × 2 cell. Hence, The nearest-neighbor exchange parameterJ0 of the local magnetic moments can be evaluated using the Ising model without an external field,  , where
, where ![[m with combining circumflex]](https://www.rsc.org/images/entities/i_char_006d_0302.gif) i and j are the local magnetic moments at the nearest-neighbor sites i and j. By comparing the DFT total energies of FM and AFM phase, the value of J0 is extracted as 6.66 meV using the following formula: J0 = ΔE/16m2. We then estimated the Curie temperature (TC) from a Monte Carlo (MC) simulation within the Ising Hamiltonian. Starting from the initial spin arrangements of FM (see Fig. 3d) in a 100 × 100 supercell containing 104 local magnetic moments, we got the variation of average energy and magnetic moment as a function of temperature shown in Fig. 3e. It can be seen that the average energy gradually rises with the increase of temperature and the magnetic moment per unit cell rapidly falls to 0.0 μB at the temperature of TC = 830 K, indicating the system undergoes a transition from FM to paramagnetic (PM) at this temperature. Although this MC simulation only gives a rough estimation of the Curie temperature, it is consistent with the high stability of the FM state superior to the AFM state. This is related to the large split between the two spin channels in the region near the Fermi level. The partially-occupied acceptor levels allow virtual hopping for the FM arrangement,27 as shown in Fig. 3c, whereas the AFM arrangement is highly energetically disadvantageous.
i and j are the local magnetic moments at the nearest-neighbor sites i and j. By comparing the DFT total energies of FM and AFM phase, the value of J0 is extracted as 6.66 meV using the following formula: J0 = ΔE/16m2. We then estimated the Curie temperature (TC) from a Monte Carlo (MC) simulation within the Ising Hamiltonian. Starting from the initial spin arrangements of FM (see Fig. 3d) in a 100 × 100 supercell containing 104 local magnetic moments, we got the variation of average energy and magnetic moment as a function of temperature shown in Fig. 3e. It can be seen that the average energy gradually rises with the increase of temperature and the magnetic moment per unit cell rapidly falls to 0.0 μB at the temperature of TC = 830 K, indicating the system undergoes a transition from FM to paramagnetic (PM) at this temperature. Although this MC simulation only gives a rough estimation of the Curie temperature, it is consistent with the high stability of the FM state superior to the AFM state. This is related to the large split between the two spin channels in the region near the Fermi level. The partially-occupied acceptor levels allow virtual hopping for the FM arrangement,27 as shown in Fig. 3c, whereas the AFM arrangement is highly energetically disadvantageous.
We now go back to the band structures of the ferromagnetic N-doped graphene kagome nanomesh and investigate its topological properties. In addition to the atomic orbitals discussed earlier in this article, the isosurfaces of Kohn–Sham wave functions shown in Fig. 2b also suggests the kagome-like bands come mainly from the marginal N and center C atoms and minor from the remaining atoms. Based on the features, we adopted a single-orbital tight-binding (TB) model with an exchange field and onsite energy to reproduce the spin-polarized kagome-like bands. In this model, we only consider the nearest-neighbor (NN) hopping between marginal N and center C atoms, leading to an extended ruby lattice, as shown in Fig. 4a. According to this approximation, the Hamiltonian is expressed as follows:

 | ||
| Fig. 4 (a) Schematic representation of the extended ruby lattice. t1, t2, t3, and t4 represent the nearest-neighbor hopping parameters and λ is the next-nearest-neighbor intrinsic SOC parameter. (b) A comparison between the band structures calculated from first-principles (DFT) and the extended ruby model. | ||
Next, we investigated the SOC effects. The band structures with SOC are almost identical to those without SOC (see Fig. 2a), except a SOC band gap of 3.7 meV at K or K′ points in proximity of the Fermi level, which can be seen clearly from the magnified bands in Fig. 5a. The SOC band gap can be well described using the above extended ruby model combined with spin–orbital interaction, and the TB Hamiltonian is written as:

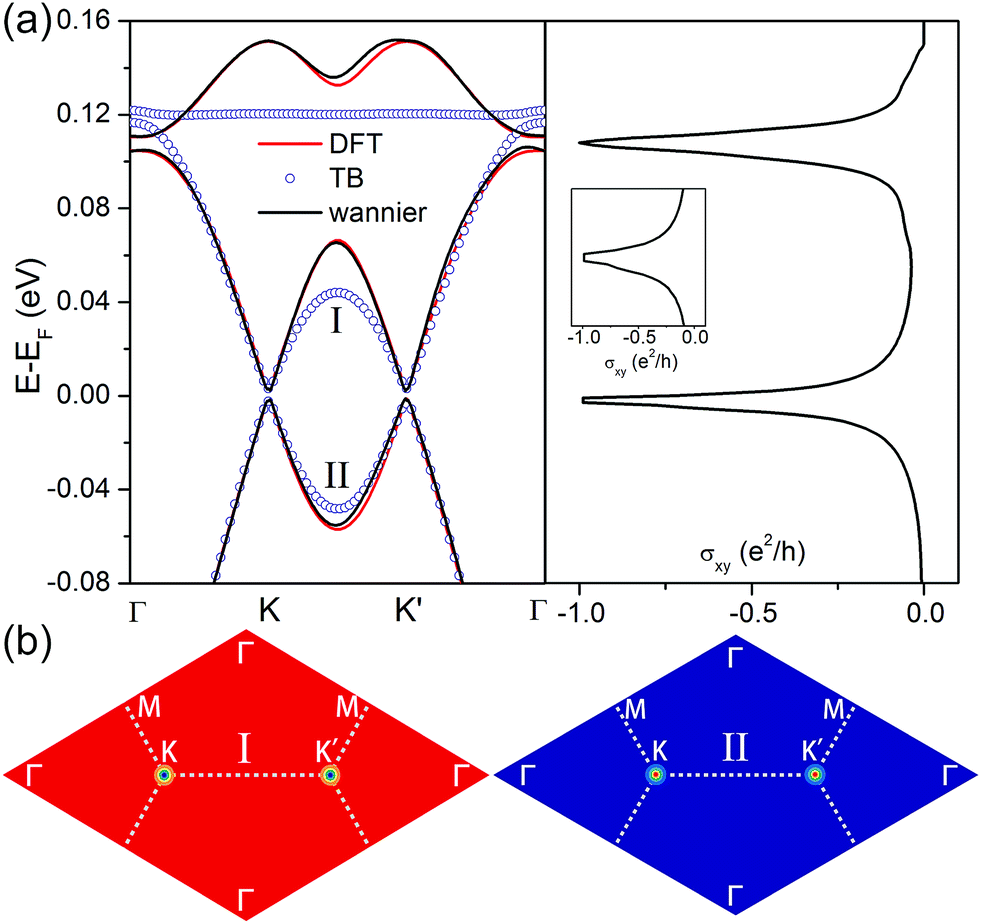 | ||
| Fig. 5 (a) Enlarged view of the kagome-like bands with SOC nearest to the Fermi level calculated from DFT, TB, and Wannier90 (left panel), and anomalous Hall conductivity calculated from the maximally localized Wannier functions (right panel). (b) The 2D distribution of the Berry curvature for the Dirac bands (I and II) obtained from TB in the momentum space. The dashed lines mark the first Brillouin zone. | ||
The spin–orbital interaction is defined as spin-dependent second-nearest-neighbor hopping. λ is spin–orbital coupling strength shown in Fig. 4a and s is the spin Pauli matrix. The unit vector êij is defined as:
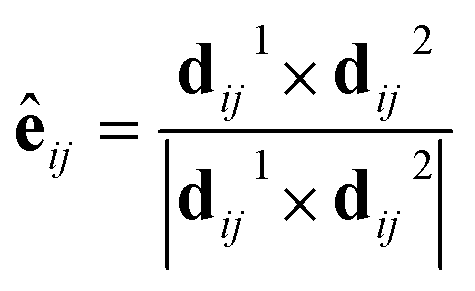
The nontrivial topology of the SOC gaps can be confirmed by the Chern number (C) calculated from the k-space integral of Barry curvature (Ω(k)) of all the states below the Fermi level using the Kubo formula:

where ψnk is the spinor Bloch wave function of band n with the corresponding eigenenergy εnk and vi is the ith Cartesian component of the velocity operator. The right panel of Fig. 5a shows the calculated anomalous Hall conductivity as a function of energy based on maximally localized Wannier functions, which develops plateaus at the value of −1.0e2/h within the energy window of SOC gaps. This implies that the N-doped graphene kagome nanomesh has a topologically nontrivial state at the Fermi level with a Chern number of C = −1. The well match between Wannier and DFT bands shown in the left pannel of Fig. 5a ensures the accuracy of the anomalous Hall conductivity. Based on the extended ruby model, we also calculated the distribution of Berry curvatures for the lowest conduction band (I) and highest valence band (II) around the Fermi level, respectively, and plotted tham in Fig. 5b. It is obvious that the Berry curvature of the two Dirac bands resides mainly around the K or K′ point in BZ. More importantly, the nontrivial SOC gap in the graphene nanomesh is well separated from other bands and thus implementable in achieving the QAHE at low temperature (∼43 K).53

Conclusions
Using first-principles calculations combined with a tight-binding model, we propose a nitrogen-doped graphene kagome nanomesh which has electronic structures characterized by kagome-like bands composing of one flat band above two Dirac bands. A moderate band gap of 3.7 meV is opened due to intrinsic SOC at the Fermi level. Anomalous Hall conductivity and Berry curvatures calculations show that the graphene nanomesh is a two-dimensional magnetic toplogical insulator characterized by a Chern number of −1, which may be implementable for achieving the QAHE at low temperature (∼43 K). The physical origins of QAHE are the relatively strong SOC effect and stable FM state provided by pxy atomic orbitals. These features can be described by using an extended ruby model. This work suggests a promising approach to search for metal-free magnetic TIs for realizing QAHE at high temperatures.Acknowledgements
This work is supported by the National Basic Research Program of China (no. 2012CB932302), the National Natural Science Foundation of China (nos 91221101 and 21433006), the 111 project (no. B13209), the Taishan Scholar Program of Shandong, and the National Super Computing Centre in Jinan.References
- F. D. M. Haldane, Phys. Rev. Lett., 1988, 61, 2015–2018 CrossRef CAS.
- M. Onoda and N. Nagaosa, Phys. Rev. Lett., 2003, 90, 206601 CrossRef.
- H. Zhang, C. Lazo, S. Blügel, S. Heinze and Y. Mokrousov, Phys. Rev. Lett., 2012, 108, 056802 CrossRef.
- H. Zhang, F. Freimuth, G. Bihlmayer, M. Ležaić, S. Blügel and Y. Mokrousov, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 205132 CrossRef.
- Z. F. Wang, Z. Liu and F. Liu, Phys. Rev. Lett., 2013, 110, 196801 CrossRef CAS.
- Y. Zhang and J. Shi, Phys. Rev. Lett., 2014, 113, 016801 CrossRef.
- R. Yu, W. Zhang, H. J. Zhang, S. C. Zhang, X. Dai and Z. Fang, Science, 2010, 329, 61–64 CrossRef CAS PubMed.
- C.-X. Liu, X.-L. Qi, X. Dai, Z. Fang and S.-C. Zhang, Phys. Rev. Lett., 2008, 101, 146802 CrossRef.
- K. Garrity and D. Vanderbilt, Phys. Rev. Lett., 2013, 110, 116802 CrossRef.
- Y. Ma, Y. Dai, X. Li, Q. Sun and B. Huang, Carbon, 2014, 73, 382–388 CrossRef CAS PubMed.
- W.-K. Tse, Z. Qiao, Y. Yao, A. H. MacDonald and Q. Niu, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155447 CrossRef.
- Z. Qiao, W. Ren, H. Chen, L. Bellaiche, Z. Zhang, A. H. MacDonald and Q. Niu, Phys. Rev. Lett., 2014, 112, 116404 CrossRef.
- Z. Qiao, S. Yang, W. Feng, W.-K. Tse, J. Ding, Y. Yao, J. Wang and Q. Niu, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 161414 CrossRef.
- H. Pan, Z. Li, C.-C. Liu, G. Zhu, Z. Qiao and Y. Yao, Phys. Rev. Lett., 2014, 112, 106802 CrossRef.
- X.-L. Zhang, L.-F. Liu and W.-M. Liu, Sci. Rep., 2013, 3, 2908 Search PubMed.
- M. Zhao, A. Wang and X. Zhang, Nanoscale, 2013, 5, 10404–10408 RSC.
- Z. Y. Zhang, J Phys Condens Matter, 2011, 23, 365801 CrossRef PubMed.
- C. Z. Chang, J. Zhang, X. Feng, J. Shen, Z. Zhang, M. Guo, K. Li, Y. Ou, P. Wei, L. L. Wang, Z. Q. Ji, Y. Feng, S. Ji, X. Chen, J. Jia, X. Dai, Z. Fang, S. C. Zhang, K. He, Y. Wang, L. Lu, X. C. Ma and Q. K. Xue, Science, 2013, 340, 167–170 CrossRef CAS PubMed.
- C. L. Kane and E. J. Mele, Phys. Rev. Lett., 2005, 95, 146802 CrossRef CAS.
- C.-C. Liu, W. Feng and Y. Yao, Phys. Rev. Lett., 2011, 107, 076802 CrossRef.
- M. Zhao and R. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 195427 CrossRef.
- M. Zhao, W. Dong and A. Wang, Sci. Rep., 2013, 3, 3532 Search PubMed.
- A. Wang, X. Zhang and M. Zhao, Nanoscale, 2014, 6, 11157–11162 RSC.
- A. Du, S. Sanvito and S. C. Smith, Phys. Rev. Lett., 2012, 108, 197207 CrossRef.
- X. Zhang, M. Zhao, A. Wang, X. Wang and A. Du, J. Mater. Chem. C, 2013, 1, 6265 RSC.
- X. Li, J. Zhou, Q. Wang, Y. Kawazoe and P. Jena, J. Phys. Chem. Lett., 2013, 4, 259–263 CrossRef CAS.
- M. W. Zhao, F. C. Pan and L. M. Mei, Appl. Phys. Lett., 2010, 96, 012508 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- K. Parlinski, Software PHONON, Cracow Search PubMed.
- A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari, Comput. Phys. Commun., 2008, 178, 685–699 CrossRef CAS PubMed.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335 RSC.
- Y. Li, Z. Zhou, P. Shen and Z. Chen, ACS Nano, 2009, 3, 1952–1958 CrossRef CAS PubMed.
- G. Ning, Z. Fan, G. Wang, J. Gao, W. Qian and F. Wei, Chem. Commun., 2011, 47, 5976–5978 RSC.
- Z. Fan, Q. Zhao, T. Li, J. Yan, Y. Ren, J. Feng and T. Wei, Carbon, 2012, 50, 1699–1703 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- J. Bai, X. Zhong, S. Jiang, Y. Huang and X. Duan, Nat. Nanotechnol., 2010, 5, 190–194 CrossRef CAS PubMed.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- W. Zhao, O. Höfert, K. Gotterbarm, J. F. Zhu, C. Papp and H. P. Steinrück, J. Phys. Chem. C, 2012, 116, 5062–5066 CAS.
- Q. Min, X. Zhang, X. Chen, S. Li and J. J. Zhu, Anal. Chem., 2014, 86, 9122–9130 CrossRef CAS PubMed.
- P. Kuhn, A. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS PubMed.
- T. V. Tam, N. B. Trung, H. R. Kim, J. S. Chung and W. M. Choi, Sens. Actuators, B, 2014, 202, 568–573 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- T. Mu, J. Huang, Z. Liu, B. Han, Z. Li, Y. Wang, T. Jiang and H. Gao, J. Mater. Res., 2011, 19, 1736–1741 CrossRef.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- C. Wu and S. Das Sarma, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235107 CrossRef.
- E. Kan, W. Hu, C. Xiao, R. Lu, K. Deng, J. Yang and H. Su, J. Am. Chem. Soc., 2012, 134, 5718–5721 CrossRef CAS PubMed.
- Z. F. Wang, N. Su and F. Liu, Nano Lett., 2013, 13, 2842–2845 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |