
Ab initio study on the stability of N-doped ZnO under high pressure
Xiaojing Sha,
Fubo Tian,
Da Li,
Defang Duan,
Binhua Chu,
Yunxian Liu,
Bingbing Liu and
Tian Cui*
State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, People's Republic of China. E-mail: cuitian@jlu.edu.cn; Fax: +86-431-85168825; Tel: +86-431-85168825
First published on 29th January 2015
Abstract
We perform first-principles density functional theory calculations to examine the stability of nitrogen-doped wurtzite ZnO under pressure. Our calculations indicate that both the stability of the nitrogen-doped ZnO and the defect concentration increase with pressure. As the pressure increases from 0 to 9 GPa, the density of states at the Fermi level decreases, and the states have a tendency to move to lower energy levels. Electron-localization function and Bader charge analysis have been used to understand the pressure effect on the defect. Under the basic growth conditions (using ε-N2 for nitrogen atoms), the calculated formation enthalpies decrease with pressure, which suggests a rise in the defect concentration. Applying pressure has great impact on the nitrogen-doped defects, and can be used as an efficient approach to form p-type ZnO.
1 Introduction
During the past years, wurtzite-ZnO has received great attention as a promising material for electronic, ferroelectric, and optical applications. ZnO has a direct band gap of 3.3 eV and a large exciton binding energy of 60 meV, thus it has drawn particular interest for potential applications in short wavelength optoelectronic devices, where the efficient excitonic UV laser actions have been demonstrated at room temperature.1–3 N-type ZnO is available even without any intentional doping, and low-resistance n-type doping with electron concentrations as high as 1021 cm−3 has been achieved.4 On the other hand, it is very difficult to obtain p-type ZnO,5 although recently it was demonstrated that high densities of holes could be obtained with N or As as the dopant using novel doping techniques.6–9The properties of semiconductors are often controlled by defects and impurities, where the incorporation of impurities in small concentrations usually affects the electrical conductivity in a significant way. Nitrogen is thought to be the most suitable p-type dopant in ZnO mainly due to its similar atomic radius and electronegativity as O, and thus should readily substitute on O sites.10 Although theory suggests some difficulties to achieve a shallow acceptor level in N-doped ZnO,6,11,12 N is still considered as the most suitable dopant for its lower lattice distortion, compared with Al, Ga, and In doping.13 Several mechanisms leading to the p-type difficulties are well known: low solubility, the compensation by low-energy native defects, deep impurity level, and structural stability known as AX and DX centers.6,9,14 N-doped ZnO, the most feasible and promising candidate for p-type ZnO,6,15–17 also suffers from the low doping efficiency and the instability of substitutional nitrogen.18 Some nonequilibrium growth techniques have been used to greatly enhance the solubility of nitrogen dopants in ZnO, such as epitaxial and molecular doping (NO2, NO, etc.).19 Moreover, new advanced approaches to create more stable and shallower acceptors in ZnO are still a subject of intense research. First-principles calculations have become more and more powerful tools to help explaining and supporting experiments, as well as predicting new materials and their properties, especially at extreme conditions such as under high pressure. Some first-principles total-energy calculations suggest shallow donor levels of the oxygen vacancy VO and zinc interstitial Zni, possibly responsible for the high n-type conductivity and the equilibrium p-type doping difficulties in ZnO.20 In previous reports, Gai et al. show that the formation enthalpies of these low energy intrinsic defects decrease with pressure, indicating possible weaker effect of compensation and favorable p-type doping at pressure.21 So the pressure is an important means to change the material properties.
Here we investigate the N-doped defects in ZnO under high pressure using a first-principles pseudo potential method. To understand the pressure dependences, we examine the geometry optimization, formation enthalpies and electronic structure of the N-doped ZnO under pressure. We describe the methods used in the calculation in Section 2, and present the calculated results and discussions of the defect formation enthalpy, transition energy levels, pressure dependence of the bond length and electronic structure in Section 3. The paper is concluded with a brief summary in Section 4.
2 Computational details
In this paper all structural optimizations, electronic band structure calculations and defect formation enthalpies for ZnO were performed using the Vienna ab initio simulation package (VASP)22 utilizing the projector augmented plane-wave (PAW) method23,24 to describe the core–valence interaction. For structure optimization, the exchange correlation (XC) functional was treated within the Ceperley–Alder LDA (CA-LDA).25 All calculations were performed treating Zn 3d electrons as valence electrons. The cut-off energy set to be 550 eV, and the appropriate Monkhorst–Pack k-point mesh were used with the resolution of 2π × 0.03 Å−1 for Brillouin zone (BZ) sampling to ensure that all the enthalpy calculations were well converged to less than 1 meV, and 72-atom cubic supercell in the wurtzite structure for all defect calculations. In all calculations, the volumes are fully relaxed until the Hellmann–Feynman forces acting on the atoms become less than 0.01 eV Å−1.The LDA band-gap error is caused by the cation-d states in II–VI band. Calculations in the GW approximation which largely correct for the LDA band-gap error for ZnO, that the occupied cation-d bands shift down relative to LDA. In research of Lany and Zunger, they used LDA+U method26 to yield agreement of the d-like density of states, and found the transition energies of Vanion calculated from LDA+U supercell energies differ only slight. The formation and transition energies presented in those work are obtained using supercell total energies that are calculation in LDA.27 In our paper, we also calculate with LDA, as many other researchers in the same way.28,29
We use first-principles supercell calculations in order to determine the enthalpy (H) at different volumes as H = E + PV + qEF, where EF is the Fermi level and q is the defect charge in electron units. V = ∂H/∂P defines the formation volume and determines the pressure dependence of the formation enthalpy. Under hydrostatic pressure, the change of crystal volume depends on both the internal strain introduced by the defect and external pressure ΔVfD = ΔVT − ΔVP. ΔVT is the total volume change of the supercell containing defect under hydrostatic pressures, ΔVP is the volume change caused by external pressure, and ΔVfD is the change of ideal crystal volume caused by the defect under hydrostatic pressure. Therefore, ΔVfD should be a constant for a given defect and is a symbol of defect concentration dependent on pressure.
The formation enthalpy of dopants has an influence on the solubility and thus the carrier density. The defect equilibrium concentration c in the thermodynamic equilibrium is related to the defect formation enthalpy Hf by:
c = Nsites![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Hf/kBT) exp(−Hf/kBT)
| (1) |
| Hf(NO, q) = H(NO, q) − H(ZnO) + μO − μN + qEF | (2) |
As we know, if the system contains an excess of O, the excess O may form a bulk O precipitate. Consequently, the chemical potential of bulk O may not exceed the chemical potential of bulk O, μO ≤ μ(bulk O). Similarly, the chemical potential of Zn may not exceed that of bulk Zn, μZn ≤ μ(bulk Zn). In addition, the sum of the O and Zn chemical potentials is required to equal the chemical potential (per pair) of bulk ZnO (we use the total enthalpy of a perfect ZnO cell at T = 0 K for μZnO), μZnO = μZn + μO. And we have neglected the temperature dependence of Eg and ΔH. In our calculations, we take the effects of stress into account, so we simply take the N2 (in ε-N2) as the N source,33 and take the oxygen O8 as the O source.34
The formation enthalpy for each charged state of defect is calculated from the total enthalpy. For all the defects considered here, the defect and hole concentrations are determined by the charge neutrality condition.35 For the charged defects a homogeneous compensating background charge was added. We adjust the charge through the parameter NELECT in the code, which equals to the total number of electrons in the system plus the number of charged impurities.
The thermodynamic transition level of a defect between two different charge states q and q′, ε(q/q′), corresponds to the Fermi level at which the formation enthalpies for the two charge states equalize. In reference to the Valence-Band Maximum (VBM), the thermodynamic transition level is given as
| ε(q/q′) = (ΔHqf,VBM − ΔHq′f,VBM)/(q′ − q) | (3) |
3 Results and discussion
Before discussing the doped ZnO, we consider the structural properties of pure ZnO. ZnO mostly crystallizes in hexagonal wurtzite-type structures (B4) at ambient pressure, and it can also be stable in the cubic zinc-blende structure (B3) by experiment.36 At about 9 GPa, ZnO transits to the rock-salt structure (B1). Hence our mainly discussion is with the B4 phase under 9 GPa. However, we performed the calculations with B1 and B3 phase as a reference also. The optimized lattice parameters of pure ZnO in B4 phase are a = 3.195 Å and c = 5.167 Å, which are in an agreement with the experimental data of 3.245 Å and 5.209 Å.11Fig. 1 shows the calculated defect formation enthalpy of the nitrogen doped in ZnO (NO), under both the O-rich and Zn-rich growth conditions, at 0, 5 and 9 GPa, as a function of the Fermi energy. We examine ZnO in the B4 phase under pressures up to 9 GPa, below which the B4 structure is known to be stable. The changes in the line slopes represent different defect charged states, while the corresponding transition levels are marked with vertical lines. The defect formation enthalpies depend strongly on the Fermi level for the charged states. NO has the lower formation enthalpies under the Zn-rich condition, than under the O-rich condition. Janotti et al. previously reported that oxygen vacancies have the lowest formation enthalpies and act as the dominant defect species under Zn-rich condition.37 Thus, less energy is generally required for N atom to enter into the oxygen vacancy sites. As shown in Fig. 1, in N-doped ZnO where the Fermi level is high in the band gap, the negatively charged cation vacancy has lower formation enthalpy which decreases with pressure, but the pressure dependence is less significant than the neutral part. Under both Zn-rich and O-rich conditions, the defect formation enthalpies decrease with the pressure increasing. That indicates that with the pressure increasing, the structure is becoming more stable.
 | ||
| Fig. 1 The defect formation enthalpies of N-doped ZnO, at both Zn-rich (left) and O-rich (right) conditions, as a function of the Fermi level. | ||
The defect formation volumes obtained via equation and the formation volumes of NO under 0, 5, and 9 GPa are −0.64, −0.63, and −0.59 Ωf.u./e (Ωf.u. denotes the volume per formula unit),38 respectively. The defect formation volume of NO is always negative under different pressures. Thus, according to V = ∂H/∂P, the formation enthalpy would decrease with pressure in principle, which contrasts our calculations in formation enthalpy.
Based on the defect formation energies, the equilibrium defect concentrations were estimated by the eqn (1). The defect concentrations of NO at different temperatures and pressures can be read from Fig. 2. We consider has the ZnO annealed at high temperature, typically 1300 K.39 At high temperatures, the defects can diffuse sufficiently rapidly that their concentration reaches equilibrium. For example, if materials were synthesized at 1300 K, the NO concentrations under 0, 5, 9 GPa would be 2.79 × 1016, 1.13 × 1017, and 1.39 × 1017 cm−3, respectively. The calculated concentration in ZnO is in good agreement with the reported value of ∼1 × 1017 cm−3.40 As Fig. 2 shown, the equilibrium concentration of NO will increase when the pressure increases.
 | ||
| Fig. 2 The concentration of defects as a function of synthesis temperature at pressure of 0, 5, and 9 GPa. | ||
The electron-localization function (ELF) of NO was calculated under 0, 5 and 9 GPa. At 0 GPa, as shown in Fig. 3, significant charge density contours can be seen between the Zn and the O atoms, as well as the Zn and N atoms, and the ionic bonding usually prevails for ZnO. After compared the ELFs between 0 and 9 GPa, we find there is very little differences between the bonds and the charge density, which can be also seen from the respective values of Bader charges at different pressures, as shown in Table 1. At atomic state, the charges of Zn and N atoms are 12 and 5 respectively. With pressure increasing, N atoms accept more charges from Zn atoms, with the charge number raised from 6.37, 6.38 to 6.39, indicating stronger ionic bonds between Zn and N. For 0 to 9 GPa is a lower pressure range, the change of charge is inconspicuous. The Bader charges has been calculated with the B3 phase, in which phase the atoms have the same coordination number with B4 phase. From 10, 30, and 60 GPa, the charge number is 6.42, 6.45, and 6.50, respectively. From the large pressure range, there is an obvious change of charge under pressure between Zn and N. So, the bond between Zn and N is stronger, when the pressure is increasing.
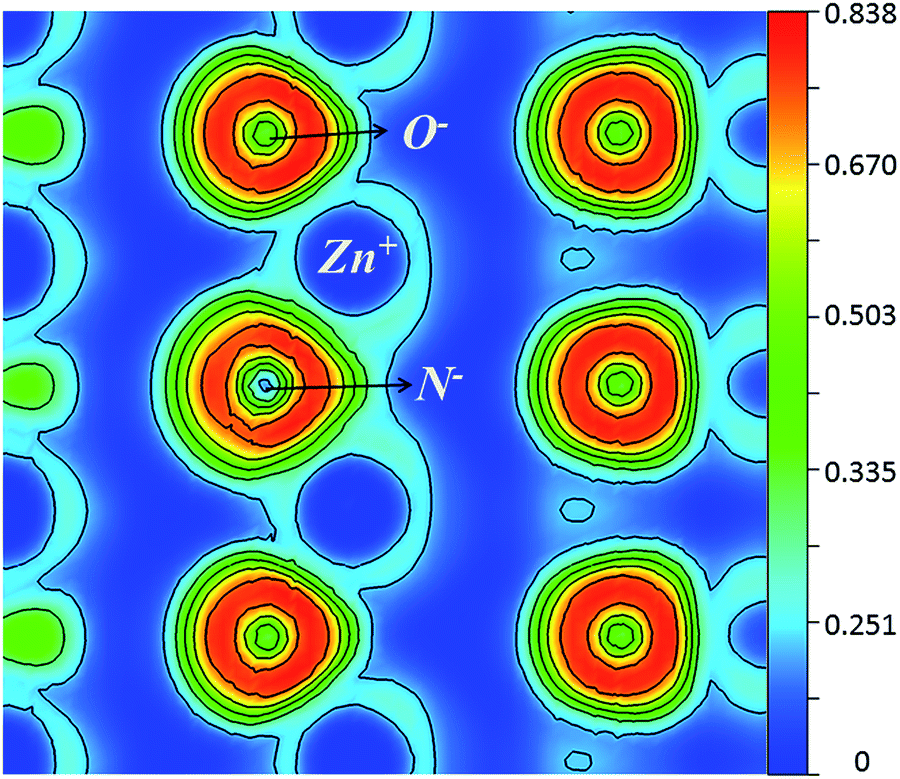 | ||
| Fig. 3 Electron localization function contour map for ZnO:N. | ||
| 0 GPa | 5 GPa | 9 GPa | |
|---|---|---|---|
| N | 1.37 | 1.38 | 1.39 |
| Zn1 | −1.26 | −1.27 | −1.27 |
| Zn2 | −1.26 | −1.27 | −1.27 |
| Zn3 | −1.26 | −1.27 | −1.28 |
| Zn4 | −1.20 | −1.21 | −1.22 |
We examine both the undoped and N-doped ZnO using supercell approach. Fig. 4 shows the calculated total density of states (DOS) for the bulk ZnO host (dash line) and N-doped ZnO (full line) at ambient pressure, in which the zero energy is set at the Fermi level. The Fermi energy, EF, is defined as the highest energy value of an occupied state in the system inside the first Brillouin zone.37 The calculated band gap of undoped ZnO is 0.9 eV. The values of the band gap calculated using the LDA are generally underestimated, in comparison to the experimental data (3.2 eV, ref. 41). As shown in Fig. 4, a defect band is generated at the top of the valence band for p-type N-doped ZnO, comparing to the DOS of undoped ZnO. For the p-type ZnO:N, we further examine the pressure dependence of the electronic structure form the partial DOS (PDOS), as shown in Fig. 5. The electronic structure of the valence band at the Fermi level are mostly from 2p states of N atoms, Zn 3p states and Zn 3d states. Those states are hybridized and the hybridization effect becomes further profound with the increase in pressure. The PDOS are calculated under different pressures (P = 0, 5 and 9 GPa). The corresponding values of DOS at the Fermi level are about 33.33, 28.78 and 27.35 states per eV per cell, respectively. The decrease in the DOS value at the Fermi surface and the shift of the impurities peaks to the lower energy both make the system more stable under pressure.42 The decreased defect formation enthalpies at pressure is previously shown to be related to the stronger bond energy from the structure property, which can be further understood from the electronic structure perspective here: the system become more stable as the hybridization effect becomes further profound with the increase in pressure, and the value of DOS at Fermi level decrease.
 | ||
| Fig. 4 Total DOS and partial DOS (PDOS) of N-doped ZnO. The Fermi energy is set to zero. | ||
 | ||
| Fig. 5 Partial DOS of 3d and 2p states of zinc, 2p state of nitrogen under 0 GPa (a) and 9 GPa (b). | ||
The calculated formation enthalpies for NO in ZnO are shown in Fig. 1. NO is stable in both the neutral and −1 charge states, with the acceptor level (0/−1) at 0.2 eV, consistent with previous results.43 The calculated acceptor level (0/−1) using hybrid functional (1.3 eV) by Lyons et al., is significantly larger than our LDA results, which might be attributed to a noticeable downward shift of the VBM on an absolute energy scale. The transition levels remain unchanged under pressure, in both Zn-rich and O-rich situations, as shown Fig. 1, indicating that pressure has little influence on the transition levels.
From the previous study,44 the formation enthalpies of VO, ZnO, and Zni increased with pressure increased, and the formation enthalpies of VZn, OZn, and Oi decreased with pressure increased. In the investigation of cation vacancies in GaN, AlN, and GaAs,45 they found the formation enthalpies of cation vacancies in AlN, GaN, and GaAs decreased with pressure increased. To analysis the effect of different ions substitution, we also discuss the situation of Na substituted for Zn. In our calculation, the formation enthalpy of NaZn increased with pressure, which is opposite with the results of NO. The phenomenon shows the cation substitutional defect becomes unstable with pressure is same with the native defect of VO, ZnO, and Zni, indicated that the change of formation enthalpy under pressure is influenced by the defect type.
4 Summary
We have studied the stability of N-doped ZnO under pressure via first-principles DFT calculations. Introducing high pressure results in a decrease in the formation enthalpy, and an increase in the equilibrium concentration of NO. The calculated values of defect formation volumes are always negative, indicated the formation enthalpy would decrease with pressure. With the increasing pressure, the impurities energy level located at Fermi energy are weakened. The Bader charge analysis shows that pressure also makes bond energy stronger. Overall, pressure provides an efficient way to approach p-type ZnO.Acknowledgements
This work was supported by the National Basic Research Program of China (no. 2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (no. IRT1132), National Natural Science Foundation of China (nos 51032001, 11074090, 11104102, 10979001, 51025206, 11204100), and National Fund for Fostering Talents of basic Science (no. J1103202). Parts of the calculations were performed at the High Performance Computing Center (HPCC) of Jilin University.References
- D. Bagnall, Y. Chen, Z. Zhu, T. Yao, S. Koyama, M. Y. Shen and T. Goto, Appl. Phys. Lett., 1997, 70, 2230–2232 CrossRef CAS PubMed.
- Z. K. Tang, G. K. L. Wong, P. Yu, M. Kawasaki, A. Ohtomo, H. Koinuma and Y. Segawa, Appl. Phys. Lett., 1998, 72, 3270–3272 CrossRef CAS PubMed.
- U. Ozgur, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Dogan, V. Avrutin, S. J. Cho and H. Morkoc, J. Appl. Phys., 2005, 98, 103 CrossRef PubMed.
- T. Minami, H. Sato, H. Nanto and S. Takata, Jpn. J. Appl. Phys., 1985, 24, L781 CrossRef.
- S. B. Zhang, S.-H. Wei and A. Zunger, J. Appl. Phys., 1988, 83, 3192–3196 CrossRef PubMed.
- C. Park, S. Zhang and S.-H. Wei, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 073202 CrossRef.
- M. Joseph, H. Tabata and T. Kawai, Jpn. J. Appl. Phys., 1999, 38, L1205 CrossRef CAS.
- Y. R. Ryu, S. Zhu, D. C. Look, J. M. Wrobel, H. M. Jeong and H. W. White, J. Cryst. Growth, 2000, 216, 330–334 CrossRef CAS.
- Y. F. Yan, S. B. Zhang and S. T. Pantelides, Phys. Rev. Lett., 2001, 86, 5723 CrossRef CAS.
- D. W. Jenkins and J. D. Dow, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 3317 CrossRef CAS.
- J. L. Lyons, A. Janotti and C. G. Van de Walle, Appl. Phys. Lett., 2009, 95, 252105 CrossRef PubMed.
- Q. Wang, Q. Sun and P. Jena, New J. Phys., 2009, 11, 14 Search PubMed.
- A. Mohanta, J. G. Simmons, H. O. Everitt, G. Shen, S. M. Kim and P. Kung, J. Lumin., 2014, 146, 470–474 CrossRef CAS PubMed.
- S. B. Zhang, J. Phys.: Condens. Matter, 2002, 14, R881 CrossRef CAS.
- A. Tsukazaki, A. Ohtomo, T. Onuma, M. Ohtani, T. Makino, M. Sumiya, K. Ohtani, S. F. Chichibu, S. Fuke and Y. Segawa, Nat. Mater., 2004, 4, 42–46 CrossRef.
- Y. Nakano, T. Morikawa, T. Ohwaki and Y. Taga, Appl. Phys. Lett., 2006, 88, 3 Search PubMed.
- L. Shen, R. Q. Wu, H. Pan, G. W. Peng, M. Yang, Z. D. Sha and Y. P. Feng, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 4 Search PubMed.
- L. Wang and A. Zunger, Phys. Rev. Lett., 2003, 90, 256401 CrossRef CAS.
- H. Matsui, H. Saeki, T. Kawai, H. Tabata and B. Mizobuchi, J. Appl. Phys., 2004, 95, 5882–5888 CrossRef CAS PubMed.
- L. Liu, J. Xu, D. Wang, M. Jiang, S. Wang, B. Li, Z. Zhang, D. Zhao, C.-X. Shan and B. Yao, Phys. Rev. Lett., 2012, 108, 215501 CrossRef.
- Y. Gai, B. Yao, Y. Li, Y. Lu, D. Shen, J. Zhang, D. Zhao, X. Fan and T. Cui, Phys. Lett. A, 2008, 372, 5077–5082 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467 CrossRef CAS.
- S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 035215 CrossRef.
- R. Vidya, P. Ravindran, H. Fjellvåg, B. G. Svensson, E. Monakhov, M. Ganchenkova and R. M. Nieminen, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 045206 CrossRef.
- F. Oba, S. R. Nishitani, S. Isotani, H. Adachi and I. Tanaka, J. Appl. Phys., 2001, 90, 824 CrossRef CAS PubMed.
- S. Zhang and J. E. Northrup, Phys. Rev. Lett., 1991, 67, 2339 CrossRef CAS.
- D. B. Laks, C. G. Van de Walle, G. F. Neumark and S. T. Pantelides, Phys. Rev. Lett., 1991, 66, 648 CrossRef CAS.
- S. B. Zhang and J. E. Northup, Phys. Rev. Lett., 1991, 67, 2339 CrossRef CAS.
- X. Wang, Y. Wang, M. Miao, X. Zhong, J. Lv, T. Cui, J. Li, L. Chen, C. J. Pickard and Y. Ma, Phys. Rev. Lett., 2012, 109, 175502 CrossRef.
- L. Zhu, Z. W. Wang, Y. C. Wang, G. T. Zou, H. Mao and Y. M. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 751 CrossRef CAS PubMed.
- J. E. Northrup and S. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 6791 CrossRef CAS.
- C. Klingshirn, Phys. Status Solidi B, 2007, 244, 3027 CrossRef CAS.
- A. Janotti and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165202 CrossRef.
- P. Erhart, K. Albe and A. Klein, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205203 CrossRef.
- G. D. Mahan, J. Appl. Phys., 1983, 54, 3825 CrossRef CAS PubMed.
- D. C. Look, D. C. Reynolds, C. W. Litton, R. L. Jones, D. B. Eason and G. Cantwell, Appl. Phys. Lett., 2002, 81, 1830 CrossRef CAS PubMed.
- S. Pearton, D. Norton, K. Ip, Y. Heo and T. Steiner, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2004, 22, 932–948 CrossRef CAS.
- P. Zhang, B. T. Wang, C. H. He and P. Zhang, Comput. Mater. Sci., 2011, 50, 12 Search PubMed.
- E.-C. Lee, Y. S. Kim, Y. G. Jin and K. J. Chang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 085120 CrossRef.
- X. Sha, F. Tian, D. Li, D. Duan, B. Chu, Y. Liu, B. Liu and T. Cui, Solid State Commun., 2015, 201, 130 CrossRef CAS PubMed.
- N. E. Christensen and A. Svane, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 075210 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |