
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydration of nitriles to amides by a chitin-supported ruthenium catalyst†
Aki
Matsuoka
a,
Takahiro
Isogawa
a,
Yuna
Morioka
a,
Benjamin R.
Knappett
b,
Andrew E. H.
Wheatley
*b,
Susumu
Saito
*ac and
Hiroshi
Naka
*a
aGraduate School of Science and Research Center for Materials Science, Nagoya University, Chikusa, Nagoya 464-8602, Japan. E-mail: h_naka@nagoya-u.jp
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: aehw2@cam.ac.uk
cInstitute for Advanced Research, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: saito.susumu@f.mbox.nagoya-u.ac.jp
First published on 8th January 2015
Abstract
Chitin-supported ruthenium (Ru/chitin) promotes the hydration of nitriles to carboxamides under aqueous conditions. The nitrile hydration can be performed on a gram-scale and is compatible with the presence of various functional groups including olefins, aldehydes, carboxylic esters and nitro and benzyloxycarbonyl groups. The Ru/chitin catalyst is easily prepared from commercially available chitin, ruthenium(III) chloride and sodium borohydride. Analysis of Ru/chitin by high-resolution transmission electron microscopy indicates the presence of ruthenium nanoparticles on the chitin support.
Introduction
The catalytic hydration of nitriles (RCN) to carboxamides (RCONH2) represents a fundamentally important pathway to these products in both laboratory and industrial contexts.1–3 Since the discovery of alumina-supported ruthenium hydroxide catalysts [Ru(OH)x/Al2O3] by Yamaguchi et al.,4 solid-supported Ru has become an important class of catalyst for nitrile hydration, demonstrating high selectivity for carboxamide formation as well as other practical advantages.5–8 Although RuCl3·nH2O itself catalyzes nitrile hydration, the choice of solid support is critically important for achieving sufficient reactivity as well as for retaining Ru species on support.4,6 Examples of supports successfully used for Ru species include inorganic γ-Al2O3,4 nanoferrite5a and magnetic silica,5b as well as organic chitosan,5c amberlite6 and Nafion.7 However, these systems typically require the use of microwave irradiation5,6 or high reaction temperatures (∼175 °C).7 Moreover, the tolerance of base-sensitive functional groups such as carboxylic esters has not been documented in these reports.4–7 Such chemoselectivity is important in modern organic synthesis,9 but is generally considered elusive in nitrile hydration promoted by metal-loaded heterogeneous catalysts, a single exception (Au/TiO2)8f notwithstanding. In this work we establish that chitin-supported ruthenium (abbreviated as Ru/chitin) serves as a versatile catalyst for the hydration of nitriles to carboxamides (Scheme 1). Using this system, nitrile hydration can be operated under near-neutral, aqueous conditions without requiring any special apparatus. Moreover, the morphologies of ruthenium nanoparticles on the chitin support were clarified by high-resolution transmission electron microscopy (HRTEM) analysis. | ||
| Scheme 1 Hydration of nitriles to carboxamides with Ru/chitin. | ||
After cellulose, chitin is the second most abundant polysaccharide in nature.10 It has a wide range of applications in materials, food, medical and environmental contexts. These include in the preparation of chitosan, affinity chromatography, wound-dressing and metal-extraction in water purification.11 Whereas chitin has been intensively used as a catalyst support for enzymes,12 its use as a support for metal catalysts has been less widely explored than has that of chitosan.13,14 So far, chitin has been used as a support for Pt in asymmetric arene hydrogenation,15 Pd in the hydrogenation of nitrobenzene and unsaturated fatty acid esters16 and Re in the epoxidation of olefins.14d We expected that chitin would represent a potentially attractive support for the Ru-catalyzed hydration of nitriles because it is highly stable under aqueous conditions and effectively adsorbs Ru species using its carboxamide functionality.17
Results and discussion
Catalytic tests
Ru/chitin catalyst was prepared by impregnating commercially available chitin with an aqueous solution of RuCl3·3H2O followed by reduction with NaBH4,18 and was tested for its effectiveness in the hydration of benzonitrile (1a, Table 1). When a mixture of 1a (1.0 mmol), H2O (1.0 mL) and Ru/chitin (0.016 mmol Ru, 1.6 mol% Ru) was heated at 120 °C for 3 h, the corresponding amide 2a was obtained in 33% 1H NMR yield (Table 1, entry 1). The presence of ruthenium was found to be essential, with the reaction hardly proceeding without catalyst or using only chitin (entries 2 and 3). Meanwhile, the chitin support was also found to be critical, with RuCl3·3H2O alone catalyzing the hydration of 1a but with significantly lower efficiency (entry 4). The optimization of reaction conditions using Ru/chitin increased the yield of 2a from 33% to 87% (entries 5–7). Ru/chitin with a higher Ru content [2.3 mol% Ru, prepared from catalyst precursor (202 mg, 1.2 wt% Ru)] gave slightly better yield still (entry 8). This result proved to be reproducible (1H NMR yields of separate runs: 97%, 91%, 89% and 89%). Analogously prepared Ru catalysts that utilized other polysaccharide supports such as chitosan and cellulose (abbreviated as Ru/chitosan and Ru/cellulose, respectively) were found to be less reactive than Ru/chitin (entries 9 and 10).|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol% Ru) | H2O (mL) | t/h | Yieldb (%) |
| a Conditions: 1a (1.0 mmol), H2O (1.0 mL, 56 equiv.) and catalyst [1.6 mol% Ru, prepared from catalyst precursor (202 mg, 0.8 wt% Ru)] at 120 °C under a N2 atmosphere unless otherwise stated. The mol% Ru was confirmed by ICP-AES analysis. b Of 2a determined by 1H NMR using mesitylene as an internal standard. c Ru/chitin (2.3 mol% Ru), prepared from catalyst precursor (1.2 wt% Ru). d Ru/chitosan (3.2 mol% Ru), prepared from catalyst precursor (1.7 wt% Ru). e Ru/cellulose (3.2 mol% Ru), prepared from catalyst precursor (1.6 wt% Ru). | ||||
| 1 | Ru/chitin (1.6) | 1 | 3 | 33 |
| 2 | None | 1 | 3 | <1 |
| 3 | Chitin | 1 | 3 | <1 |
| 4 | RuCl3·3H2O (2.0) | 1 | 3 | 17 |
| 5 | Ru/chitin (1.6) | 1 | 20 | 77 |
| 6 | Ru/chitin (1.6) | 1 | 30 | 87 |
| 7 | Ru/chitin (1.6) | 4 | 20 | 87 |
| 8 | Ru/chitin (2.3)c | 4 | 20 | 97 |
| 9 | Ru/chitosan (3.2)d | 4 | 20 | 74 |
| 10 | Ru/cellulose (3.2)e | 4 | 20 | 17 |

Scope and limitation
The scope of the Ru/chitin-catalyzed hydration of nitriles is outlined in Table 2. These reactions were run under comparable conditions to those in entry 8 of Table 1. Benzamide (2a) was obtained in 87% isolated yield (Table 2, entry 1) and variously substituted benzonitriles could be converted to the corresponding amides in good-to-excellent yield (entries 2–13). o-Methyl-substituted 1i was somewhat less reactive (entry 9) though m- and p-substituted analogues reacted in satisfying yields (entries 7 and 8). Benzonitrile 1l, which bore an electron-withdrawing p-nitro group, was completely hydrated in shorter reaction times than 1b, 1c, 1f and 1g, each of which bore electron-donating groups at the para positions. p-Formylbenzonitrile (1k) could be converted to the corresponding amide 2k with an intact formyl moiety in 76% yield, though formation of the hydrate of the aldehyde was also noted under aqueous conditions. Furthermore, heteroaromatic nitriles could be efficiently hydrated to the corresponding amides (entries 14 and 15).| Entry | Nitrile (1) | t/h | Amide (2) | Isolated yield (%) | ||
|---|---|---|---|---|---|---|
| a Conditions: 1 (1.0 mmol), H2O (4 mL, 220 equiv.) and Ru/chitin (2.3 mol% Ru) at 120 °C under a N2 atmosphere. | ||||||
| 1 |

|
1a | 20 |

|
2a | 87 |
| 2 |

|
1b | 60 |

|
2b | 89 |
| 3 |

|
1c | 40 |

|
2c | 98 |
| 4 |
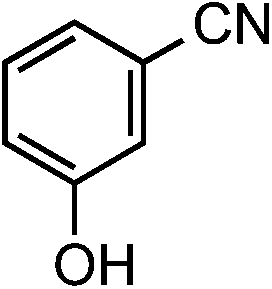
|
1d | 32 |

|
2d | 97 |
| 5 |

|
1e | 32 |

|
2e | 79 |
| 6 |

|
1f | 40 |
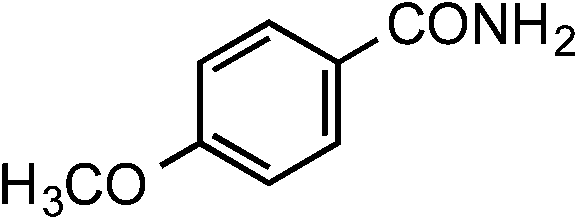
|
2f | 88 |
| 7 |

|
1g | 50 |

|
2g | 96 |
| 8 |

|
1h | 60 |

|
2h | 92 |
| 9 |

|
1i | 60 |

|
2i | 49 |
| 10 |

|
1j | 24 |

|
2j | 80 |
| 11 |

|
1k | 40 |

|
2k | 76 |
| 12 |

|
1l | 18 |

|
2l | 92 |
| 13 |

|
1m | 50 |

|
2m | 65 |
| 14 |

|
1n | 24 |

|
2n | 94 |
| 15 |

|
1o | 20 |

|
2o | 91 |
| 16 | CH3CN | 1p | 36 | CH3CONH2 | 2p | 65 |
| 17 |

|
1q | 36 |

|
2q | 55 |
| 18 |

|
1r | 48 |

|
2r | 65 |
| 19 |
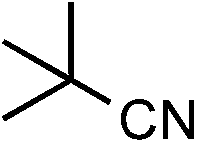
|
1s | 48 |

|
2s | 23 |
| 20 |

|
1t | 36 |

|
2t | 85 |
| 21 |

|
1u | 24 |

|
2u | 66 |
| 22 |

|
1v | 30 |
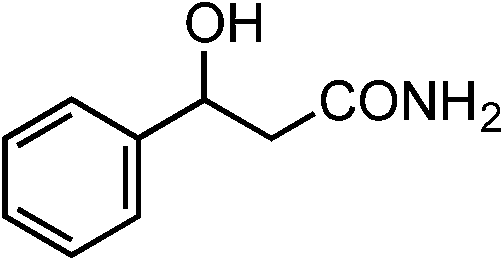
|
2v | 46 |
| 23 |

|
1w | 24 |

|
2w | 81 |
The Ru/chitin system was also applied to the hydration of aliphatic nitriles (Table 2, entries 16–23). Although the hydration reaction proved susceptible to steric hindrance (entry 19), primary and secondary nitriles 1p–r and 1t–w could all be converted to amides (entries 16–18 and 20–23) with retention of olefin (entry 21), β-hydroxy (entry 22) and α-methoxy (entry 23) groups in fair-to-good yields.
Importantly, the presence of a base-sensitive carbonyl functionality in methyl ester 1x was tolerated by virtue of the near-neutral conditions that could be used for catalyst preparation6,18 (Scheme 2). Similarly, an α-amino nitrile conjugated with a redox-sensitive benzyloxycarbonyl (Cbz) group (as in 1y) was converted to protected α-amino amide 2y with retention of the carbamoyl linkage. The tolerance to carboxylic ester and CbzN functionality shown in Scheme 2 illustrates the applicability of the present method to the nitrile hydration of complex molecules bearing redox- or base-sensitive functional groups.
 | ||
| Scheme 2 Hydration nitriles bearing carboxylic ester and carbamate functionalities. | ||
Upscale experiment
To verify the scalability of this method, a gram-scale hydration was carried out (Scheme 3). The hydration of 1w using a decreased loading of Ru/chitin catalyst (0.5 mol% Ru) yielded the amide 2w in excellent yield [conditions: 1w (16 mmol), H2O (13 mL), Ru/chitin (0.5 mol% Ru), 120 °C, 36 h, 92% isolated yield (77% isolated yield after 24 h under otherwise identical conditions)]. | ||
| Scheme 3 Gram-scale hydration of nitrile 1w. | ||
HRTEM analysis
To elucidate the nature of the Ru/chitin catalyst, the catalyst was analyzed by HRTEM. The presence of nanoparticles with a mean size of 2.1 ± 0.4 nm was established (Fig. 1a–c). Energy dispersive X-ray spectroscopy (EDX) and measurement of the d-spacings (d = 0.23 nm) indicated the presence of both Ru(0) and RuO2 [Fig. 1c, inset, and Fig. 1d]. EDX also revealed the presence of Ca and P in both Ru/chitin (Fig. 1d) and chitin (Fig. 1e and f). This was attributed to calcium phosphate on account of the crustaceous origin of the chitin and was found not to incur significant catalytic activity (Table 1, entry 3). | ||
| Fig. 1 (a) A histogram representing the particle size distribution of Ru nanoparticles on chitin. (b) Low-magnification and ((c) and inset) high resolution TEM images of chitin-supported Ru nanoparticles. (d) EDX of Ru/chitin. (e) A low-magnification TEM image of chitin. (f) EDX of chitin. | ||
TEM analysis of the Ru nanoparticles after hydration of 1w showed that they remained morphologically essentially unchanged (Fig. 2). In fact, the Ru/chitin catalyst could be reused without significant loss of catalytic activity (hydration of 1a to 2a, conditions: identical to Table 1, entry 8, first run, 95% yield; reuse run, 87% yield).
 | ||
| Fig. 2 (a) Low-magnification and (b) high resolution TEM images of Ru/chitin after nitrile hydration. | ||
Conclusions
We have established that chitin-supported ruthenium displays high catalytic activity towards the hydration of nitriles to amides under aqueous conditions. The catalyst is easily prepared, and applicable to the hydration of a wide variety of nitriles with aromatic, heteroaromatic and aliphatic substituents. HRTEM analysis of Ru/chitin revealed the presence of ruthenium nanoparticles before and after hydration reactions, indicating that chitin could serve as an effective solid support for ruthenium nanoparticles.Experimental section
General comments
1H and 13C NMR spectra were recorded on a JEOL ECA-600 (600 MHz for 1H, 150 MHz for 13C, 564 MHz for 19F) or a JEOL ECA-500 (500 MHz for 1H, 125 MHz for 13C) at 25 °C. Chemical shifts are reported as δ in ppm and are internally referenced to tetramethylsilane (TMS, 0.00 ppm for 1H), CD2HOH (3.30 ppm for 1H), CD2HSOCD3 (2.50 ppm for 1H), HOD (4.79 ppm for 1H), CDCl3 (77.2 ppm for 13C), CD3OD (49.0 ppm for 13C), dioxane (67.2 ppm in D2O for 13C), or dimethyl sulfoxide-d6 (DMSO-d6, 39.5 ppm for 13C). Chemical shifts for 19F NMR were externally referenced to CF3COOH (−78.5 ppm, neat). Infrared (IR) spectra were recorded on a FT-IR6100 (JASCO). High-resolution mass spectrometry (HRMS) was recorded with a Bruker Daltonik micrOTOF-QII spectrometer. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) spectra were recorded on an Agilent VISTA-PRO. Elemental analyses were recorded on a Yanaco CHN recorder MT-6. These analytical experiments were carried out at the Chemical Instrumental Center, Research Center for Materials Science, Nagoya University. Melting points were recorded on an OptiMelt automated melting point system (Stanford Research Systems). Ruthenium content was analyzed by ICP-AES using yttrium as an internal standard after digestion of samples (10 mg) in concd HNO3 (2 mL) at 150 °C for 12 h. Products 2a–y were known compounds and their identities were confirmed by comparing with literature data.6,7,8g,19–34TEM analysis
High-resolution transmission electron microscopy (HRTEM) analysis was performed on a JEOL JEM-3011 microscope. Samples illustrated in Fig. 1a–d and 2 were prepared as per the typical procedure for the preparation of Ru/chitin (0.016 mmol Ru, vide infra) and by the hydration of 1w (0.5 mmol) using Ru/chitin (0.016 mmol Ru) at 120 °C for 6 h, respectively. Sample preparation required droplet coating of particle dispersions obtained by sonicating in CH3CH2OH on carbon-coated Cu grids (Agar Scientific, 300 mesh). Electron optical parameters: CS = 0.6 mm, CC = 1.2 mm, electron energy spread = 1.5 eV, beam divergence semi-angle = 1 mrad. Elemental analysis was by energy dispersive X-ray spectroscopy (EDX) using a PGT prism Si/Li detector and an Avalon 2000 analytical system. Spectra were analyzed using the PGT eXcalibur 4.03.00 software. Observed Cu Kα and Kβ emission lines were attributed to scattered electrons impinging on the copper grid. Any minor Fe Kα and Co Kα emission lines of similar intensity were due to parasitic scattering from the lens polepiece. Detailed analysis of particle morphology was performed using Digital Micrograph 3.6.5 by counting the diameters of 100 particles (N), defining intervals of 0.25 nm between dmin ≤ d ≤ dmax and counting the number of particles falling into these intervals. Particle size distributions were constructed using DataGraph 3.0. Values of average d-spacing were obtained from Fourier transforms of high magnification images (×800k, ×1 M) using d = 20/D where D is the diameter (nm) of rings obtained. Average d-spacing was confirmed using the profile tool in Digital Micrograph by averaging over 10 d-spacings. To determine the error in the value of d-spacing thus obtained, detailed TEM examination of CeO2 and Au nanoparticles was undertaken. The relationship between FT ring diameter and DV value (a measure of objective lens focusing voltage) was established for DV values between −6 and +6 and the standard deviation in d-spacing was established to be 10% when compared to the literature.Materials
RuCl3·3H2O was purchased from Furuya Metal Co., Ltd. Benzonitrile (1a), m-hydroxybenzonitrile (1d), p-methylbenzonitrile (1g), m-methylbenzonitrile (1h), o-methylbenzonitrile (1i), m-chlorobenzonitrile (1j), p-nitrobenzonitrile (1l), 2,3,4,5,6-pentafluorobenzonitrile (1m), acetonitrile (1p), pivalonitrile (1s), 2-phenylacetonitrile (1t), 3-hydroxy-3-phenylpropanonitrile (1v), α-methoxyacetonitrile (1w) and 4-(methoxycarbonyl)benzonitrile (1x), were purchased from TCI. p-Aminobenzonitrile (1b), p-methoxybenzonitrile (1f), 2-furonitrile (1o), yttrium standard solution [Y(NO3)3 in HNO3, 1.00 mg Y per mL; 1000 ppm] and concd HNO3 were purchased from Wako Chemicals. p-Hydroxybenzonitrile (1c), p-formylbenzonitrile (1k), propionitrile (1q), 2-methylpropanenitrile (1r) and acrylonitrile (1u) were purchased from Aldrich. o-Hydroxybenzonitrile (1e) was purchased from Merck. Nicotinonitrile (1n) and chitin were purchased from Kanto Chemicals. Ruthenium standard solution (1 mg Ru per mL in HCl) for ICP-AES was purchased from Acros. α-(Benzyloxycarbonylamino)acetonitrile (1y) was prepared according to the literature procedure by protecting α-aminoacetonitrile with CbzCl.35Catalyst preparation
Hydration of nitriles to amides
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7; tetrahydrofuran/diethyl ether 1:10) to afford amide 2a as colorless plates (103.8 mg, 87% yield). Mp 126.5–127.1 °C (lit.19 125–128 °C); IR (KBr) 1405, 1577, 1625, 1660, 3175, 3369 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.36 (bs, 1H), 7.42–7.47 (m, 2H), 7.51 (tt, J = 1.5, 7.3 Hz, 1H), 7.85–7.89 (m, 2H), 7.97 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 127.4, 128.2, 131.2, 134.3, 167.9; elemental analysis calcd for [C7H7NO·0.1H2O]: C, 68.39; H, 5.90; N, 11.39, found: C, 68.64; H, 5.92; N, 11.15.
:8); mp 176.3–180.9 °C (lit.19 180–183 °C); IR (KBr) 780, 1397, 1563, 1600, 3219, 3326, 3464 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 5.59 (s, 2H), 6.53 (d, J = 8.6 Hz, 2H), 6.86 (bs, 1H), 7.53 (bs, 1H), 7.60 (d, J = 8.6 Hz, 2H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 112.5, 121.0, 129.2, 151.7, 168.2; elemental analysis calcd for [C7H8N2O]: C, 61.75; H, 5.92; N, 20.58, found: C, 61.79; H, 5.87; N, 20.30.
:6–10:0, gradient); mp 157.1–159.2 °C (lit.20 148 °C); IR (KBr) 1247, 1403, 1558, 1618, 1649, 3124, 3340, 3417 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 6.78 (d, J = 8.6 Hz, 2H), 7.08 (bs, 1H), 7.68–7.79 (m, 3H), 9.94 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 114.7, 125.0, 129.5, 160.2, 167.7; elemental analysis calcd for [C7H7NO2]: C, 61.31; H, 5.15; N, 10.21, found: C, 61.13; H, 5.14; N, 10.01.
:20:200); mp 165.1–168.0 °C (lit.21 167–168 °C); IR (KBr) 1256, 1450, 1580, 1653, 3247, 3400 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 6.88–6.92 (m, 1H), 7.20–7.24 (m, 1H), 7.24–7.30 (m, 3H), 7.86 (s, 1H), 9.60 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 114.5, 118.0, 118.1, 129.2, 135.8, 157.3, 168.0; elemental analysis calcd for [C7H7NO2]: C, 61.31; H, 5.15; N, 10.21, found: C, 61.23; H, 5.12; N, 10.05.
:10–10:0, gradient); mp 164.3–169.3 °C; IR (KBr) 1146, 1253, 1396, 1422, 1573, 1643, 3170, 3391 cm−1; 1H NMR (600 MHz, CD3OD) δ 3.82 (s, 3H), 6.93–6.97 (m, 2H), 7.81–7.85 (m, 2H); 13C{1H} NMR (150 MHz, CD3OD) δ 55.9, 114.7, 126.9, 130.6, 164.1, 172.0; elemental analysis calcd for [C8H9NO2]: C, 63.56; H, 6.00; N, 9.27, found: C, 63.37; H, 6.00; N, 9.23.
:5); mp 157.6–159.0 °C (lit.8g 159–160 °C); IR (KBr) 1397, 1414, 1571, 1618, 1671, 3168, 3343 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.34 (s, 3H), 7.22–7.30 (m, 3H), 7.75–7.80 (m, 2H), 7.89 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 20.9, 127.5, 128.7, 131.5, 141.0, 167.8; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 70.99; H, 6.66; N, 10.26.
:3); mp 91.0–92.4 °C (lit.8g 92–93 °C); IR (KBr) 687, 1114, 1387, 1432, 1616, 1650, 3195, 3376 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.34 (s, 3H), 7.25–7.35 (m, 3H), 7.64–7.69 (m, 1H), 7.69–7.72 (m, 1H), 7.91 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 20.9, 124.6, 128.1 (2C), 131.7, 134.3, 137.4, 168.0; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 71.04; H, 6.78; N, 9.98.
:3); mp 140.1–140.5 °C (lit.8g 140 °C); IR (KBr) 1140, 1395, 1623, 1656, 3187, 3368 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.36 (s, 3H), 7.18–7.24 (m, 2H), 7.28–7.37 (m, 3H), 7.68 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 19.6, 125.4, 127.0, 129.1, 130.4, 135.1, 137.1, 171.0; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 71.35; H, 6.72; N, 10.36.
:1; ethyl acetate only; tetrahydrofuran only) and activated carbon treatment; mp 132.6–133.4 °C (lit.8g 134–136 °C); IR (KBr) 1123, 1389, 1432, 1569, 1626, 1658, 3181, 3366 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.46–7.52 (m, 1H), 7.54 (bs, 1H), 7.56–7.61 (m, 1H), 7.81–7.86 (m, 1H), 7.89–7.94 (m, 1H), 8.10 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 126.2, 127.3, 130.3, 131.1, 133.1, 136.3, 166.4; elemental analysis calcd for [C7H6NClO]: C, 54.04; H, 3.89; N, 9.00, found: C, 53.96; H, 3.89; N, 8.95.
:3); mp 155.4–159.2 °C; IR (KBr) 1212, 1397, 1607, 1670, 1698, 3194, 3288, 3354 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.60 (bs, 1H), 7.96–8.00 (m, 2H), 8.03–8.08 (m, 2H), 8.17 (bs, 1H), 10.08 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 128.1, 129.3, 137.8, 139.3, 167.0, 192.9; elemental analysis calcd for [C8H7NO2]: C, 64.42; H, 4.73; N, 9.39, found: C, 64.69; H, 4.85; N, 9.16.
:3:7); mp 197.1–200.0 °C; IR (KBr) 1346, 1413, 1526, 1600, 1678, 3177, 3477 cm−1; 1H NMR (600 MHz, CD3OD) δ 8.05–8.08 (m, 2H), 8.29–8.33 (m, 2H); 13C{1H} NMR (150 MHz, CD3OD) δ 124.6, 130.0, 140.9, 151.2, 170.1; elemental analysis calcd for [C7H6N2O3]: C, 50.61; H, 3.64; N, 16.86, found: C, 50.67; H, 3.69; N, 16.64.
:2); mp 146.8–147.2 °C; IR (KBr) 1001, 1493, 1675, 3186, 3358 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 8.16 (bs, 1H), 8.30 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 112.9 (t, J = 21.6 Hz), 135.9–137.9 (m), 139.9–141.9 (m), 141.8–143.9 (m), 158.3; 13C{19F} NMR (150 MHz, DMSO-d6); δ 112.9 (d, J = 8.7 Hz), 136.9, 140.9, 142.9, 158.3; 19F NMR (564 MHz, DMSO-d6) δ −162.0–161.3 (m, 2F), −154.0–153.4 (m, 1F), −142.3–141.9 (m, 2F); elemental analysis calcd for [C7H2F5NO]: C, 39.83; H, 0.96; N, 6.64, found: C, 40.00; H, 6.65; N, 1.09.
:4) and recrystallization (from water); mp 139.6–140.9 °C (lit.24 141–142 °C); IR (KBr) 1626, 1665, 3171, 3351 cm−1; 1H NMR (600 MHz, CDCl3) δ 5.53 (bs, 1H), 6.26 (bs, 1H), 6.52–6.53 (m, 1H), 7.16–7.18 (m, 1H), 7.46–7.49 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 112.5, 115.3, 144.6, 147.6, 160.4; elemental analysis calcd for [C5H5NO2]: C, 54.05; H, 4.54; N, 12.61, found: C, 53.73; H, 4.55; N, 12.49.
:6–0:7, gradient); mp 121.0–121.7 °C; IR (KBr) 1624, 1658, 3194, 3386 cm−1; 1H NMR (600 MHz, CD3OD) δ 2.52 (dd, J = 4.8, 14.4 Hz, 1H), 2.63 (dd, J = 8.9, 14.5 Hz, 1H), 5.05 (dd, J = 4.8, 8.9 Hz, 1H), 7.22–7.39 (m, 5H); 13C{1H} NMR (125 MHz, CD3OD) δ 46.1, 71.9, 126.9, 128.5, 129.4, 145.4, 176.4; elemental analysis calcd for [C9H11NO2]: C, 65.44; H, 6.71; N, 8.48, found: C, 65.41; H, 6.71; N, 8.45.
:4); mp 205.0–206.6 °C (lit.33 200–201 °C); IR (KBr) 1281, 1660, 1725, 3195, 3406 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 3.87 (s, 3H), 7.57 (bs, 1H), 7.96–8.04 (m, 4H), 8.15 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 52.3, 127.8, 129.1, 131.8, 138.4, 165.7, 167.0; elemental analysis calcd for [C9H9NO3]: C, 60.33; H, 5.06; N, 7.82, found: C, 60.32; H, 5.14; N, 7.72.
:5–92:8, gradient); mp 135.0–136.6 °C (lit.34 138–139 °C); IR (KBr) 1537, 1652, 1688, 3191, 3324, 3381 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 3.55 (d, J = 6.2 Hz, 2H), 5.03 (s, 2H), 7.01 (s, 1H), 7.26–7.40 (m, 7H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 43.3, 65.4, 127.7, 127.8, 128.3, 137.1, 156.4, 171.1; HRMS (ESI-TOF) m/z calcd for C10H12N2O3Na [M + Na]+ 231.0746, found 231.0757.
:7; tetrahydrofuran/diethyl ether 1:10) to afford amide 2a as colorless plates (103.8 mg, 87% yield). Mp 126.5–127.1 °C (lit.19 125–128 °C); IR (KBr) 1405, 1577, 1625, 1660, 3175, 3369 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.36 (bs, 1H), 7.42–7.47 (m, 2H), 7.51 (tt, J = 1.5, 7.3 Hz, 1H), 7.85–7.89 (m, 2H), 7.97 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 127.4, 128.2, 131.2, 134.3, 167.9; elemental analysis calcd for [C7H7NO·0.1H2O]: C, 68.39; H, 5.90; N, 11.39, found: C, 68.64; H, 5.92; N, 11.15.
:8); mp 176.3–180.9 °C (lit.19 180–183 °C); IR (KBr) 780, 1397, 1563, 1600, 3219, 3326, 3464 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 5.59 (s, 2H), 6.53 (d, J = 8.6 Hz, 2H), 6.86 (bs, 1H), 7.53 (bs, 1H), 7.60 (d, J = 8.6 Hz, 2H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 112.5, 121.0, 129.2, 151.7, 168.2; elemental analysis calcd for [C7H8N2O]: C, 61.75; H, 5.92; N, 20.58, found: C, 61.79; H, 5.87; N, 20.30.
:6–10:0, gradient); mp 157.1–159.2 °C (lit.20 148 °C); IR (KBr) 1247, 1403, 1558, 1618, 1649, 3124, 3340, 3417 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 6.78 (d, J = 8.6 Hz, 2H), 7.08 (bs, 1H), 7.68–7.79 (m, 3H), 9.94 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 114.7, 125.0, 129.5, 160.2, 167.7; elemental analysis calcd for [C7H7NO2]: C, 61.31; H, 5.15; N, 10.21, found: C, 61.13; H, 5.14; N, 10.01.
:20:200); mp 165.1–168.0 °C (lit.21 167–168 °C); IR (KBr) 1256, 1450, 1580, 1653, 3247, 3400 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 6.88–6.92 (m, 1H), 7.20–7.24 (m, 1H), 7.24–7.30 (m, 3H), 7.86 (s, 1H), 9.60 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 114.5, 118.0, 118.1, 129.2, 135.8, 157.3, 168.0; elemental analysis calcd for [C7H7NO2]: C, 61.31; H, 5.15; N, 10.21, found: C, 61.23; H, 5.12; N, 10.05.
:10–10:0, gradient); mp 164.3–169.3 °C; IR (KBr) 1146, 1253, 1396, 1422, 1573, 1643, 3170, 3391 cm−1; 1H NMR (600 MHz, CD3OD) δ 3.82 (s, 3H), 6.93–6.97 (m, 2H), 7.81–7.85 (m, 2H); 13C{1H} NMR (150 MHz, CD3OD) δ 55.9, 114.7, 126.9, 130.6, 164.1, 172.0; elemental analysis calcd for [C8H9NO2]: C, 63.56; H, 6.00; N, 9.27, found: C, 63.37; H, 6.00; N, 9.23.
:5); mp 157.6–159.0 °C (lit.8g 159–160 °C); IR (KBr) 1397, 1414, 1571, 1618, 1671, 3168, 3343 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.34 (s, 3H), 7.22–7.30 (m, 3H), 7.75–7.80 (m, 2H), 7.89 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 20.9, 127.5, 128.7, 131.5, 141.0, 167.8; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 70.99; H, 6.66; N, 10.26.
:3); mp 91.0–92.4 °C (lit.8g 92–93 °C); IR (KBr) 687, 1114, 1387, 1432, 1616, 1650, 3195, 3376 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.34 (s, 3H), 7.25–7.35 (m, 3H), 7.64–7.69 (m, 1H), 7.69–7.72 (m, 1H), 7.91 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 20.9, 124.6, 128.1 (2C), 131.7, 134.3, 137.4, 168.0; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 71.04; H, 6.78; N, 9.98.
:3); mp 140.1–140.5 °C (lit.8g 140 °C); IR (KBr) 1140, 1395, 1623, 1656, 3187, 3368 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 2.36 (s, 3H), 7.18–7.24 (m, 2H), 7.28–7.37 (m, 3H), 7.68 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 19.6, 125.4, 127.0, 129.1, 130.4, 135.1, 137.1, 171.0; elemental analysis calcd for [C8H9NO]: C, 71.09; H, 6.71; N, 10.36, found: C, 71.35; H, 6.72; N, 10.36.
:1; ethyl acetate only; tetrahydrofuran only) and activated carbon treatment; mp 132.6–133.4 °C (lit.8g 134–136 °C); IR (KBr) 1123, 1389, 1432, 1569, 1626, 1658, 3181, 3366 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.46–7.52 (m, 1H), 7.54 (bs, 1H), 7.56–7.61 (m, 1H), 7.81–7.86 (m, 1H), 7.89–7.94 (m, 1H), 8.10 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 126.2, 127.3, 130.3, 131.1, 133.1, 136.3, 166.4; elemental analysis calcd for [C7H6NClO]: C, 54.04; H, 3.89; N, 9.00, found: C, 53.96; H, 3.89; N, 8.95.
:3); mp 155.4–159.2 °C; IR (KBr) 1212, 1397, 1607, 1670, 1698, 3194, 3288, 3354 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 7.60 (bs, 1H), 7.96–8.00 (m, 2H), 8.03–8.08 (m, 2H), 8.17 (bs, 1H), 10.08 (s, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 128.1, 129.3, 137.8, 139.3, 167.0, 192.9; elemental analysis calcd for [C8H7NO2]: C, 64.42; H, 4.73; N, 9.39, found: C, 64.69; H, 4.85; N, 9.16.
:3:7); mp 197.1–200.0 °C; IR (KBr) 1346, 1413, 1526, 1600, 1678, 3177, 3477 cm−1; 1H NMR (600 MHz, CD3OD) δ 8.05–8.08 (m, 2H), 8.29–8.33 (m, 2H); 13C{1H} NMR (150 MHz, CD3OD) δ 124.6, 130.0, 140.9, 151.2, 170.1; elemental analysis calcd for [C7H6N2O3]: C, 50.61; H, 3.64; N, 16.86, found: C, 50.67; H, 3.69; N, 16.64.
:2); mp 146.8–147.2 °C; IR (KBr) 1001, 1493, 1675, 3186, 3358 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 8.16 (bs, 1H), 8.30 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 112.9 (t, J = 21.6 Hz), 135.9–137.9 (m), 139.9–141.9 (m), 141.8–143.9 (m), 158.3; 13C{19F} NMR (150 MHz, DMSO-d6); δ 112.9 (d, J = 8.7 Hz), 136.9, 140.9, 142.9, 158.3; 19F NMR (564 MHz, DMSO-d6) δ −162.0–161.3 (m, 2F), −154.0–153.4 (m, 1F), −142.3–141.9 (m, 2F); elemental analysis calcd for [C7H2F5NO]: C, 39.83; H, 0.96; N, 6.64, found: C, 40.00; H, 6.65; N, 1.09.
:4) and recrystallization (from water); mp 139.6–140.9 °C (lit.24 141–142 °C); IR (KBr) 1626, 1665, 3171, 3351 cm−1; 1H NMR (600 MHz, CDCl3) δ 5.53 (bs, 1H), 6.26 (bs, 1H), 6.52–6.53 (m, 1H), 7.16–7.18 (m, 1H), 7.46–7.49 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 112.5, 115.3, 144.6, 147.6, 160.4; elemental analysis calcd for [C5H5NO2]: C, 54.05; H, 4.54; N, 12.61, found: C, 53.73; H, 4.55; N, 12.49.
:6–0:7, gradient); mp 121.0–121.7 °C; IR (KBr) 1624, 1658, 3194, 3386 cm−1; 1H NMR (600 MHz, CD3OD) δ 2.52 (dd, J = 4.8, 14.4 Hz, 1H), 2.63 (dd, J = 8.9, 14.5 Hz, 1H), 5.05 (dd, J = 4.8, 8.9 Hz, 1H), 7.22–7.39 (m, 5H); 13C{1H} NMR (125 MHz, CD3OD) δ 46.1, 71.9, 126.9, 128.5, 129.4, 145.4, 176.4; elemental analysis calcd for [C9H11NO2]: C, 65.44; H, 6.71; N, 8.48, found: C, 65.41; H, 6.71; N, 8.45.
:4); mp 205.0–206.6 °C (lit.33 200–201 °C); IR (KBr) 1281, 1660, 1725, 3195, 3406 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 3.87 (s, 3H), 7.57 (bs, 1H), 7.96–8.04 (m, 4H), 8.15 (bs, 1H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 52.3, 127.8, 129.1, 131.8, 138.4, 165.7, 167.0; elemental analysis calcd for [C9H9NO3]: C, 60.33; H, 5.06; N, 7.82, found: C, 60.32; H, 5.14; N, 7.72.
:5–92:8, gradient); mp 135.0–136.6 °C (lit.34 138–139 °C); IR (KBr) 1537, 1652, 1688, 3191, 3324, 3381 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 3.55 (d, J = 6.2 Hz, 2H), 5.03 (s, 2H), 7.01 (s, 1H), 7.26–7.40 (m, 7H); 13C{1H} NMR (150 MHz, DMSO-d6) δ 43.3, 65.4, 127.7, 127.8, 128.3, 137.1, 156.4, 171.1; HRMS (ESI-TOF) m/z calcd for C10H12N2O3Na [M + Na]+ 231.0746, found 231.0757.
Acknowledgements
This work was partly supported by grant from Yoshida Foundation for Science and Technology (to H.N.) and the program “Integrated Research on Chemical Synthesis”, MEXT, Japan (to H.N.). A.M. acknowledges a Grant-in-Aid for JSPS fellows (25 4305) and IGER program at NU. B.R.K. thanks the UK EPSRC for financial support (EP/J500380/1) and A.E.H.W. thanks the GB Sasakawa Foundation for travel grant 4388. The authors are grateful to Drs K. Oyama and Y. Maeda (Chemical Instrument Center, NU) for technical support. The authors wish to thank Prof. R. Noyori (NU and RIKEN) for fruitful discussions.Notes and references
- (a) C. E. Mabermann, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 1, pp. 251–266 Search PubMed; (b) D. Lipp, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 1, pp. 266–287 Search PubMed; (c) R. Opsahl, in Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz, Wiley, New York, 1991, vol. 2, pp. 346–356 Search PubMed.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (b) C. Singh, V. Kumar, U. Sharma, N. Kumar and B. Singh, Curr. Org. Synth., 2013, 10, 241–264 CrossRef CAS.
- (a) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS PubMed; (b) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS PubMed; (c) T. Tu, Z. Wang, Z. Liu, X. Feng and Q. Wang, Green Chem., 2012, 14, 921–924 RSC; (d) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC; (e) E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28–37 CrossRef CAS PubMed.
- K. Yamaguchi, M. Matsushita and N. Mizuno, Angew. Chem., Int. Ed., 2004, 43, 1576–1580 CrossRef CAS PubMed.
- (a) V. Polshettiwar and R. S. Varma, Chem.–Eur. J., 2009, 15, 1582–1586 CrossRef CAS PubMed; (b) R. B. N. Baig and R. S. Varma, Chem. Commun., 2012, 48, 6220–6222 RSC; (c) R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122–2127 RSC.
- S. Kumar and P. Das, New J. Chem., 2013, 37, 2987–2990 RSC.
- G. K. S. Prakash, S. B. Munoz, A. Papp, K. Masood, I. Bychinskaya, T. Mathew and G. A. Olah, Asian J. Org. Chem., 2012, 1, 146–149 CrossRef CAS.
- Recent examples based on other metals: (a) A. Ishizuka, Y. Nakazaki and T. Oshiki, Chem. Lett., 2009, 38, 360–361 CrossRef CAS; (b) T. Mitsudome, Y. Mikami, H. Mori, S. Arita, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2009, 3258–3260 RSC; (c) S. Ichikawa, S. Miyazoe and O. Matsuoka, Chem. Lett., 2011, 40, 512–514 CrossRef CAS; (d) M. Tamura, H. Wakasugi, K.-i. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428–11431 CrossRef CAS PubMed; (e) A. Y. Kim, H. S. Bae, S. Park, S. Park and K. H. Park, Catal. Lett., 2011, 141, 685–690 CrossRef CAS; (f) Y.-M. Liu, L. He, M.-M. Wang, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 1392–1396 CrossRef CAS PubMed; (g) Y. Gangarajula and B. Gopal, Chem. Lett., 2012, 41, 101–103 CrossRef CAS; (h) K. Yamaguchi, Y. Wang, H. Kobayashi and N. Mizuno, Chem. Lett., 2012, 41, 574–576 CrossRef CAS; (i) K.-i. Shimizu, T. Kubo, A. Satsuma, T. Kamachi and K. Yoshizawa, ACS Catal., 2012, 2, 2467–2474 CrossRef CAS; (j) T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425–6433 CrossRef CAS PubMed; (k) M. B. Gawande, P. S. Branco, I. D. Nogueira, C. A. A. Ghumman, N. Bundaleski, A. Santos, O. M. N. D. Teodoro and R. Luque, Green Chem., 2013, 15, 682–689 RSC; (l) C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed.
- (a) N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262–310 CrossRef CAS PubMed; (b) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954–10990 CrossRef CAS PubMed.
- (a) Chitin: Formation and Diagenesis, ed. N. S. Gupta, Springer, New York, 2011 Search PubMed; (b) V. S. Yeul and S. S. Rayalu, J. Polym. Environ., 2013, 21, 606–614 CrossRef CAS.
- (a) M. N. V. R. Kumar, React. Funct. Polym., 2000, 46, 1–27 CrossRef CAS; (b) M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS PubMed.
- B. Krajewska, Enzyme Microb. Technol., 2004, 35, 126–139 CrossRef CAS PubMed.
- (a) E. Guibal, Prog. Polym. Sci., 2005, 30, 71–109 CrossRef CAS PubMed; (b) D. J. Macquarrie and J. J. E. Hardy, Ind. Eng. Chem. Res., 2005, 44, 8499–8520 CrossRef CAS.
- (a) F. Quignard, A. Choplin and A. Domard, Langmuir, 2000, 16, 9106–9108 CrossRef CAS; (b) V. Calò, A. Nacci, A. Monopoli, A. Fornaro, L. Sabbatini, N. Cioffi and N. Ditaranto, Organometallics, 2004, 23, 5154–5158 CrossRef; (c) A. Corma, P. Concepción, I. Domínguez, V. Fornés and M. J. Sabater, J. Catal., 2007, 251, 39–47 CrossRef CAS PubMed; (d) A. D. Giuseppe, M. Crucianelli, M. Passacantando, S. Nisi and R. Saladino, J. Catal., 2010, 276, 412–422 CrossRef PubMed; (e) A. Ricci, L. Bernardi, C. Gioia, S. Vierucci, M. Robitzer and F. Quignard, Chem. Commun., 2010, 46, 6288–6290 RSC; (f) A. C. Kathalikkattil, J. Tharun, R. Roshan, H.-G. Soek and D.-W. Park, Appl. Catal., A, 2012, 447–448, 107–114 CrossRef CAS PubMed.
- G.-L. Yuan, M.-Y. Yin, T.-T. Jiang, M.-Y. Huang and Y.-Y. Jiang, J. Mol. Catal. A: Chem., 2000, 159, 45–50 CrossRef CAS.
- (a) Y. Ito and M. Inoue, Kagaku to Kyoiku, 2010, 58, 486–489 CAS; (b) D. Abiru and M. Inoue, Kagaku to Kyoiku, 2014, 62, 36–39 CAS.
- (a) B. J. Arena, US Pat. 4274980, 1981; (b) B. J. Arena, US Pat. 4367355, 1983; (c) R. A. A. Muzzarelli, Water Res., 1970, 4, 451–455 CrossRef CAS.
- M. Zahmakıran and S. Özkar, J. Mol. Catal. A: Chem., 2006, 258, 95–103 CrossRef PubMed.
- Q. Song, Q. Feng and K. Yang, Org. Lett., 2014, 16, 624–627 CrossRef CAS PubMed.
- J. Lee, M. Kim, S. Chang and H.-Y. Lee, Org. Lett., 2009, 11, 5598–5601 CrossRef CAS PubMed.
- D. R. Brown, I. Collins, L. G. Czaplewski and D. J. Hayden, WO Patent 2007/107758, 2007.
- X.-b. Jiang, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Org. Chem., 2004, 69, 2327–2331 CrossRef CAS PubMed.
- SDBSWeb: http://sdbs.db.aist.go.jp/ (National Institute of Advanced Industrial Science and Technology, accessed August 2014) SDBS no. 553.
- L. Zhang, S. Wang, S. Zhou, G. Yang and E. Sheng, J. Org. Chem., 2006, 71, 3149–3153 CrossRef CAS PubMed.
- SDBSWeb: http://sdbs.db.aist.go.jp/ (National Institute of Advanced Industrial Science and Technology, accessed August 2014) SDBS no. 1491.
- SDBSWeb: http://sdbs.db.aist.go.jp/ (National Institute of Advanced Industrial Science and Technology, accessed August 2014) SDBS no. 1431.
- K. Y. Koltunov, S. Walspurger and J. Sommer, Eur. J. Org. Chem., 2004, 4039–4047 CrossRef CAS.
- SDBSWeb: http://sdbs.db.aist.go.jp/ (National Institute of Advanced Industrial Science and Technology, accessed August 2014) SDBS no. 4737.
- S. S. Deshmukh, S. N. Huddar, D. S. Bhalerao and K. G. Akamanchi, ARKIVOC, 2010, 2, 118–126 CrossRef.
- SDBSWeb: http://sdbs.db.aist.go.jp/ (National Institute of Advanced Industrial Science and Technology, accessed August 2014) SDBS no. 1461.
- A. Kamal, G. B. R. Khanna, T. Krishnaji and R. Ramu, Bioorg. Med. Chem. Lett., 2005, 15, 613–615 CrossRef CAS PubMed.
- K. L. Breno, M. D. Pluth and D. R. Tyler, Organometallics, 2003, 22, 1203–1211 CrossRef CAS.
- B. F. Plummer, M. Menendez and M. Songster, J. Org. Chem., 1989, 54, 718–719 CrossRef CAS.
- S.-T. Chen, S.-H. Wu and K.-T. Wang, Synthesis, 1989, 37–38 CrossRef.
- P. Chen, L. B. Horton, R. L. Mikulski, L. Deng, S. Sundriyal, T. Palzkill and Y. Song, Bioorg. Med. Chem. Lett., 2012, 22, 6229–6232 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data. See DOI: 10.1039/c4ra15682j |
| This journal is © The Royal Society of Chemistry 2015 |