
Pdots nanoparticles load photosensitizers and enhance efficiently their photodynamic effect by FRET†
Elina Haimov*a,
Hana Weitmana,
Debby Ickowiczb,
Zvi Malikb and
Benjamin Ehrenberga
aDepartment of Physics and Institute of Nanotechnology and Advanced Materials, Bar Ilan University, Ramat Gan 52900, Israel. E-mail: ehren@mail.biu.ac.il; elinhaimov@gmail.com; Fax: +972-3-7384054; Tel: +972-3-5318427
bFaculty of Life Sciences, Bar Ilan University, Ramat Gan 52900, Israel
First published on 28th January 2015
Abstract
A new type of nanoparticles, Pdots, and a new methodology of photosensitization are developed to achieve a more efficient photodynamic effect in aqueous solutions and in cells. Pdots are nano-sized particles, composed of conjugated chromophoric polymers coated with PEGylated phospholipids. They exhibit good aqueous colloidal properties, a broad absorption band and a strong and narrow emission band. We show that these characteristics improve biological photosensitization, which is employed in the photodynamic therapy of cancer. Amphiphilic photosensitizers such as Rose Bengal partition, non-covalently but with a high affinity, into the amphiphilic coating of the Pdots, without necessitating covalent attachment. At this close contact, very efficient fluorescence resonance energy transfer (FRET) occurs between the Pdot donor and the sensitizer acceptor. The Pdots serve as broad-band collectors of light, which is funneled, via energy transfer, to the photosensitizer. Therefore, energy transfer from them can additively assist in the activity of the acceptors of energy. The energy transfer mechanism, strong uptake of the Pdot-sensitizer dyads by MCF-7 adenocarcinoma cells and their enhanced photosensitized killing are demonstrated.
Introduction
The employment of various types of nanoparticles is revolutionizing biological and medical sciences. Among the important nanostructures that have found use in biology and medicine one can list quantum dots (QDs),1,2 nanoparticles of gold, silicon, silica, metal oxides and several forms of carbon.3,4 Nanoparticles are used as carriers and drug-delivery vehicles, as biosensors, as imaging tools and as markers, in which their intrinsic chemical and optical properties are employed for analytical and diagnostic purposes.Photodynamic therapy (PDT) is a novel method for treating solid tumors. It employs photosensitizer molecules, mainly tetrapyrrole derivatives, which have two important attributes: preferential uptake by solid tumors and efficient production, upon light absorption, of the electronically excited molecular oxygen, singlet oxygen, and other reactive oxygen species. Singlet oxygen is highly cytotoxic, causing oxidative destruction of the cells in which it has been generated. The clinical protocol involves intravenous injection or topical application of the sensitizer, followed by illumination with light that is absorbed by the sensitizer, either superficially or endoscopically through optical fibers for tumors in internal organs. Illumination also elicits the sensitizer's fluorescence, which is used to mark the tumor and its borders. Because of the preferential uptake of photosensitizers by malignant tissue and because the illumination is local, no harmful side effects are observed. PDT has been used for treatment of skin, lung, breast, bladder and other tumors and has also been applied to eliminate viruses and bacteria.5–12
We suggest a novel approach that is based on employing a relatively new nanoparticle, Pdot, which has hitherto not been used in cellular PDT. Pdots are composed of chromophoric organic homo-polymers. They are a fascinating new type of nanostructure, with many potential applications, whose applications have started to be pursued. Their use, mainly as biological markers, was pioneered by McNeill.13,14 They can be processed quite easily using simple methodologies, by mixing them with various amphiphilic molecules, such as PEGylated phospholipids15 or a PEGylated polystyrene backbone,16 forming by sonication, or by just mixing, very stable colloidal particles in water. When such particles are formed, the hydrophobic lipid or polystyrene backbone is most likely embedded and entangled with the chromophoric polymer, which is in the core of the Pdot, while the hydrophilic PEG-chains extend outside, into the aqueous phase. The stabilizing polymer thus forms a biocompatible coating shell, which endows the particles with water solubility that can be tuned by the structure of the copolymer, and it also enables functionalization of the Pdots. This coating forms a nano-sized environment into which small amphiphilic molecules, such as photosensitizers, might partition and intercalate. Unlike QDs, Pdots do not exhibit size-dependent absorption and emission bands, and in contrast with QDs, are non-toxic.
The most important advantage that we foresee for this application arises from the following attribute. When a photosensitizer is in close contact with a nanoparticle, such as Pdot, that exhibits broad and strong absorption in the visible range, if the particle's brightness is high and the overlap between its emission spectrum and the sensitizer's absorption spectrum is good, then at close proximity, resonance non-radiative energy transfer from the particle to the sensitizer can turn into a big improvement.17 The particle will absorb over a broad range and will funnel and transduce the absorbed energy to the sensitizer. The nanoparticles will thus bring about that a broader spectral range, beyond the sensitizer's own narrow, and in many cases, weak absorption, could be utilized for the photochemical reaction. Pdots would thus serve as a tool to perform two aims: to load and carry active photosensitizers for treatment and tumor demarcation in PDT, but mainly to enhance their activity by serving as a light-collecting antenna. This will overcome the sensitizer's marginal intrinsic light absorption in the far red range, which is the practical spectral range for use in tissues. FRET from the nanoparticle donor will occur in addition to the sensitizer's intrinsic direct absorption, so it will undoubtedly enhance the efficiency of the photochemical reaction that underlies PDT.
Experimental
Materials
We employed the conjugated polymer poly[(9,9-dioctylfluorenyl-2,7-diyl)-alt-co-(1,4-benzo-{2,1′,3}-thiadiazole)] (ADS133YE, nicknamed PF; MW 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000), purchased from American Dye Source (Quebec, Canada), for the preparation of Pdots. The following phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA): 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-350] (PEG350-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-2000] (PEG2000-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-5000] (PEG5000-PE) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC). Rose Bengal (RB) was obtained from BDH Merck Ltd., Poole, Dorset, UK and RB Methyl Ester (RBME) was from Spectra Group Limited, Inc., Millbury, OH. The singlet oxygen chemical quencher 9, 10-dimethylanthracene (DMA) was purchased from Sigma-Aldrich (St. Louis, MO). Tetrahydrofuran was from Bio-Lab (Jerusalem, Israel) and dichloromethane was from Carlo Erba Reagents (Rodano, Italy), both were of analytical grade. Propidium iodide (PI) was from MBL (Nagoya, Japan).
000–200000), purchased from American Dye Source (Quebec, Canada), for the preparation of Pdots. The following phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL, USA): 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-350] (PEG350-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethyleneglycol)-2000] (PEG2000-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-5000] (PEG5000-PE) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC). Rose Bengal (RB) was obtained from BDH Merck Ltd., Poole, Dorset, UK and RB Methyl Ester (RBME) was from Spectra Group Limited, Inc., Millbury, OH. The singlet oxygen chemical quencher 9, 10-dimethylanthracene (DMA) was purchased from Sigma-Aldrich (St. Louis, MO). Tetrahydrofuran was from Bio-Lab (Jerusalem, Israel) and dichloromethane was from Carlo Erba Reagents (Rodano, Italy), both were of analytical grade. Propidium iodide (PI) was from MBL (Nagoya, Japan).
Preparation of Pdots
1 mg of the conjugated polymer PF was placed in 20 mL of dichloromethane and was stirred for 48 hours to maximize its solubilization. The solution was transferred through a 0.2 μm membrane filter. 4, 10 or 21 mg of PEG350-PE, PEG2000-PE or PEG5000-PE, respectively, and 5 mg of DPPC were added, in order to obtain Pdots which differ in their shell's composition. The solution was stirred for 10 minutes and was then added to 25 mL of doubly-distilled water, under constant stirring. After sonication of the solution for 20 min by a Ti-probe sonicator (MSE Soniprep 150, Crawley, UK) at 4 °C it became homogeneous. The solution was paper-filtered immediately and centrifuged for 30 min at 2500 rpm. At this step the solution separated into two phases, an upper aqueous phase and a lower organic phase, which was discarded. To determine the Pdot's concentration in the aqueous suspension, we measured the amount of material that was lost along the procedure of preparation at each step. The yield of the preparation was 23%. The concentration of the Pdots' dye material in the final aqueous suspension was 0.0123 mg mL−1.Size distribution of Pdots was determined by dynamic light scattering (DLS). Analyses were performed with a Cordouan Vasco particle size analyzer set at 25 °C, with a probing laser wavelength of 657 nm and a static detector set at an angle of 135° from the incident beam. The laser intensity was set to 56%, 47% and 80% of the maximal power (65 mW) for PF-PEG350, PF-PEG2000 and PF-PEG5000, respectively. The correlator parameters, i.e. time interval and number of channels, were set to 9 μs and 400, respectively for PF-PEG350, 8 μs and 300 for PF-PEG2000, 10 μs and 400 for PF-PEG5000 Pdots. All acquisitions were made in multi-acquisition mode on 100 measurements with a time step of 50–60 s. The particle size distribution was calculated using a Padé–Laplace model, to take into account the nanoparticles' polydispersity. In order to have statistical information on size dispersion of the samples, the NanoQTM software of VASCO was operated in the multi-acquisition mode with each correlogram acquisition processed by the Padé–Laplace inversion algorithm.
Another size characterization was made by Atomic Force Microscopy (AFM). One drop of the nanoparticles Pdots dispersion was placed on a freshly cleaved silica wafer substrate. After evaporation of the water, the surface was scanned with the AFM's probe. AFM measurements and imaging were carried out using an ICON scanning probe microscope (Bruker AXS, Santa Barbara, CA). All images were obtained using the ScanAsyst mode with a single SNL (Sharp Nitride Lever) probe (force constant of 0.12–0.48 N m−1, Bruker, Camarillo, CA). The resonance frequency of this cantilever was approximately 40–75 kHz. The scan angle was maintained at 0°. The aspect ratio was 1:1 and image resolution was 1024 samples per line for PF-PEG350 and PF-PEG5000 Pdots and 384 samples per line for PF-PEG2000, respectively. Before analysis of the images, the 1st ordering “flatting” function was applied to each image. The dimensions of the PF Pdots were determined by an analysis of the height image using the NanoScop Software.
Spectroscopic measurements
Absorption spectra were recorded on a Shimadzu (Kyoto, Japan) UV-2501PC UV-visible spectrophotometer. Fluorescence excitation and emission spectra and fluorescence time-drive measurements were performed on a Perkin-Elmer LS-50B digital fluorimeter (Norwalk, CT).All samples had small absorbance, <0.05, at the fluorescence excitation wavelength, to obtain a linear dependence of fluorescence intensity on concentration. Resolution of fluorescence spectra into two Lorentzian-shaped bands was accomplished by a non-linear routine (Origin, Microcal Software, Northampton, MA).
The appropriate concentration of Rose Bengal's and Rose Bengal Methyl Ester's stock suspension for these experiments was 0.1 mM in water:THF (9:1 v/v).
Fluorescence decays were measured at 25 °C on a home-built system, which employs time-correlated single photon counting. The excitation source has a picosecond mode-locked Ti:sapphire laser (Tsunami, Spectra-Physics, Santa Clara, CA) pumped by a high-power diode laser (MilleniaXsJ, Spectra-Physics). The second harmonic, generated by the GWU, Spectra Physics, frequency doubler, was used for excitation at 445 nm. Emission was collected at 531 nm and focused at the photocathode of a micro channel plate (MCP, Hammamatsu 3809U-50) biased at −3200 V. Photon-counting was achieved with a Becker and Hickl plate (model 630). The responses of the system yielded a pulse width of 35 ps. Life-time analyses were done by the Marquardt nonlinear least-squares method.
Singlet oxygen quantum yield
We prepared aqueous suspensions of Pdots at concentrations ranges of 1.64 × 10−4–0.003 mg mL−1 and 5 μM 9,10-dimethylanthracene (DMA). The florescent DMA molecule is used as a singlet oxygen chemical trap, since it reacts selectively and rapidly with it to form the non-fluorescent 9,10-endoperoxide (DMAO2).18 A diode-pumped solid state laser (Ningbo Lasever Inc., Ningbo, China) beam at 473 nm, whose radiation wavelength is within the Pdots' absorption band, was chosen. The laser's power was measured with a Nova power meter (Ophir, Jerusalem, Israel). The photo-production rate of excited photosensitizer molecules, in molar concentration units per second, kpho, is given by the following equation:
 | (1) |
P is the laser power, in mW, abs is the absorbance at the laser's wavelength, L is the length of the laser beam path inside the sample (in cm), NA is the number of Einstein units (1 Einstein = 6.023 × 1023 photons) of light energy per second per Watt of light at the illumination wavelength and V is the sample's volume (mL). The factor 0.98 corrects for light reflection at the air/sample interface. The diminishing fluorescence intensity of DMA was measured in a time-drive mode while, simultaneously, the laser irradiated the sample along the cuvette's long axis, perpendicular to the fluorimeter's excitation and emission direction, and its light was absorbed by the Pdots. The sample was stirred magnetically throughout the illumination, to obtain uniform exposure of the whole sample to the light. The fluorescence traces of DMA were fitted to exponential decays by a least-squares fitting (Origin, Microcal Software, Northampton, MA).
The values for singlet oxygen QY were determined by statistically averaging at least three independent measurements. The weighted average, μ, of several measurements, each yielding a result Xi and possessing a measurement error σi, was calculated using the formula:19
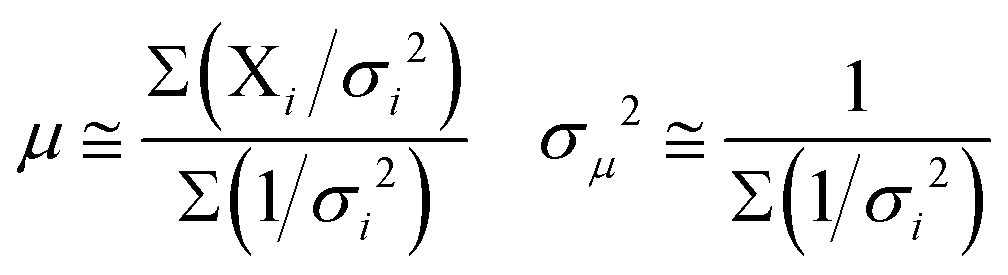 | (2) |
The uncertainty of the calculated average, σμ, is also given in eqn (2).
Cell culture
MCF-7 human breast adenocarcinoma cells were obtained from American Type Culture Collection (ATCC, Manassas, USA) and were grown on tissue culture plates (Greiner, Stroudwater, UK) in Dulbecco's Modified Eagle Medium (Sigma-Aldrich, St. Louis MO, US). The medium was enriched with 10% fetal bovine serum, 2 mM L-glutamine and penicillin (0.1 mg mL−1) (Biological Industries, Beit-Haemek, Israel). The cells were incubated at 37 °C in a humidified atmosphere with 5% CO2/95% air and were sub-cultured twice a week.Flow cytometric analyses of Pdots' uptake and cellular integrity
MCF-7 cells were incubated in serum-free DMEM medium with Pdots for up to 4 hours. The cells were then washed twice with filtered PBS and collected in the dark. Their fluorescence intensity in cells was measured by flow cytometry (FACS, Beckman Coulter Inc., CA, USA) with laser excitation at 488 nm and emission filtered at 530 nm, with 30 nm band width. Cell viability was monitored following photosensitization by either 3 × 10−5 mg mL−1 Pdots only, or by 75 nM RB only, or by both 75 nM RB and Pdots. Cells were washed twice with filtered PBS and re-suspended in 1 mL filtered PBS with 1 μM propidium iodide, a DNA-intercalating dye that is used to stain permeable cells, and collected in the dark. PI's fluorescence was measured in 104 cells per sample by FACS, with excitation at 488 nm and emission filtered at 620 nm, with 30 nm band width.Cellular fluorescence was captured and photographed using an ImageStreamX high-resolution imaging flow cytometer (Amnis, Co., Seattle, WA). Approximately 5000 cell samples were excited using a 488 nm and 561 nm lasers (200 mW). The images that were made per each cell were collected in the bright field (420–480 nm), CH2 (480–560 nm) and CH3 (560–595 nm) emission modes. Following the flow cytometry image capture, samples were gated to produce populations of captured single-cell images of living cells that were in focus. Brightness and contrast for the green and orange fluorescence intensities were adjusted similarly for each channel in the samples. The two images obtained from the two emission modes were used to quantify the co-localization of two probes, Pdot and RB, in a defined region. A co-localization parameter can thus be used to assess whether the two different “targets” are located in the same area of the cell or very near to one another, which indicates the FRET phenomena. The values for the cell analyses were determined by statistically averaging of three experiments.
Electron microscopy
Cells were harvested, washed with PBS and attached to 10 mm cover slips coated with poly-L-lysine 0.1% for 1 hour. Fixation of samples was performed according to a modification of the triple fixation GTGO method for scanning electron microscopy (SEM): the samples were fixed with 2.5% glutaraldehyde in phosphate buffer, followed by post-fixation in 2% OsO4. The third fixative was 2% tannic acid–guanidine hydrochloride. After fixation, the cells were dehydrated in graded ethanol solutions, and then the ethanol was exchanged for Freon 113-tf using graded solutions. The samples were then air-dried, gold-coated, and examined using an FEI Quanta 200FEG scanning electron microscope.20Results and discussion
The aim of this paper is to probe the possibility of using Pdots as broad-band light collectors, which transfer the absorbed energy by a resonance mechanism to a nearby photosensitizer molecule. Attempts were made to employ quantum dots as transducers of energy to photosensitizers. A successful covalent conjugation to quantum dots (QDs) was reported by Tsay et al.21 McNeill et al. reported intra-particulate energy transfer from the conjugated polymer to tetraphenyl porphyrin that was doped into the particle in its preparation, and exhibited strong one- and two-photon absorption.22 The current paper reports on ‘attaching’ photosensitizers to Pdots, without necessitating covalent binding, which could be a big advantage in the employment of these particles, since the same batch of Pdots could be used to attach different amphiphilic or lipophilic molecules. We report the very efficient energy transfer and photosensitized cancer cell-killing that were obtained by this approach.Absorption and emission spectra
The absorption and fluorescence spectra of Pdots, composed of the PF polymer, Rose Bengal and Rose Bengal Methyl Ester in aqueous solution as well as dyads made of Pdots with either RB or RBME are also shown in Fig. 1a and b respectively. It can be seen that the combination of Pdots with RB or RBME exhibits broad and strong absorption in the visible range.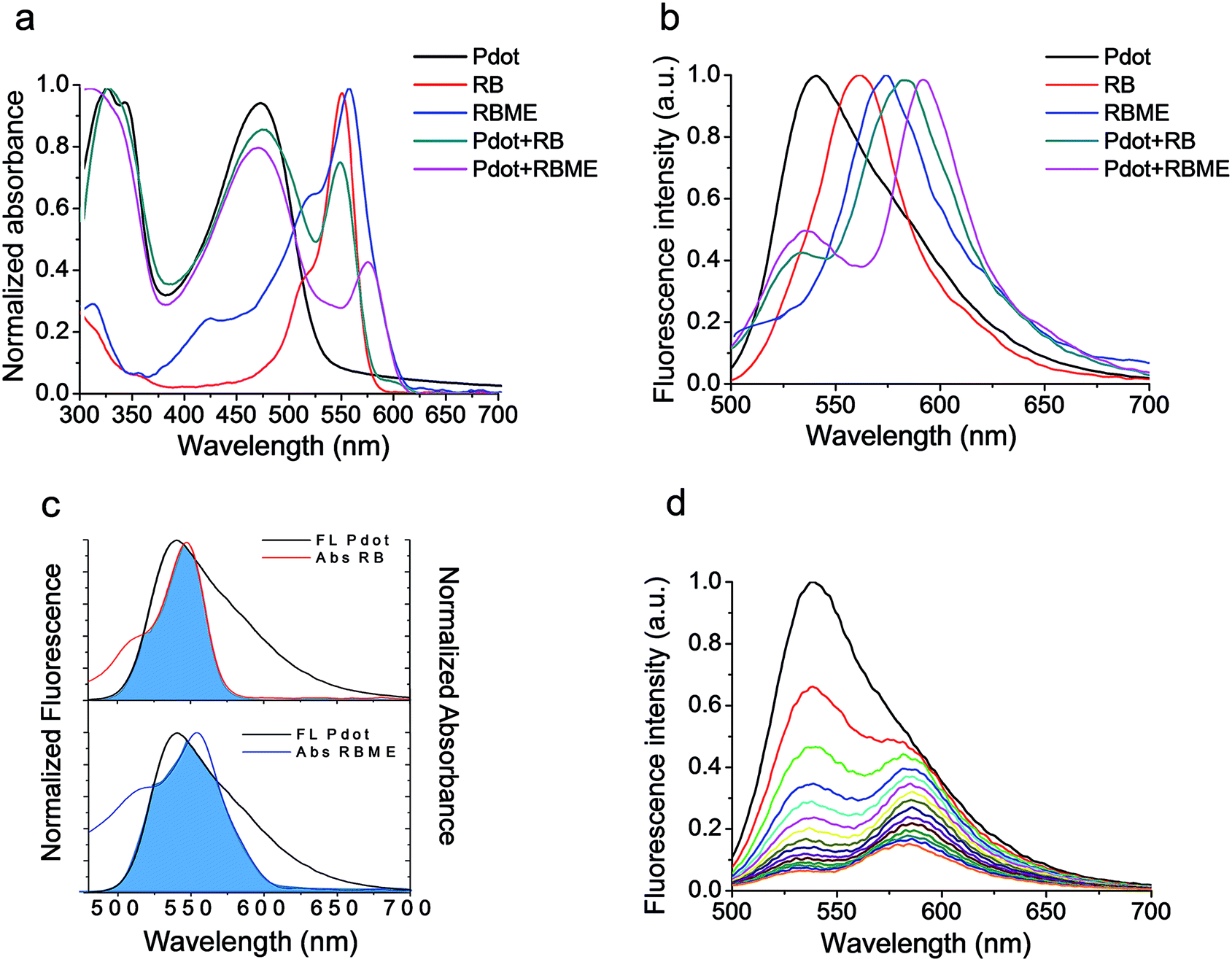 | ||
| Fig. 1 Absorbance and fluorescence spectra. (a) Normalized absorption spectra and (b) fluorescence spectra (λex = 460 nm) of PF-PEG350 Pdots, RB in water, RBME in water, PF-PEG350 Pdots with RB and PF-PEG350 Pdots with RBME. (c) Spectral overlap integral between the fluorescence spectrum of PF-PEG350 Pdots and the absorption spectra of RB (upper panel) and RBME (lower panel). (d) Fluorescence of 1.2 × 10−4 mg mL−1 PF-PEG350 Pdots with increasing concentration of RB, 0–4 μM (λex = 460 nm). | ||
We chose RB, which has been studied for its efficient photosensitization efficiency,23,24 as a sensitizer. The main absorption bands of RB and RBME have maxima at 547 nm and 555 nm, respectively. Pdots composed of PF with a coating surface consisting of PEG350 exhibit an emission maximum at 538 nm. Pdots with other PEGylated coatings had almost identical spectra. Thus, a good overlap exists between the Pdots' fluorescence spectrum and the absorption spectra of RB or RBME, seen in Fig. 1c, as required for efficient FRET.
Fig. S1† shows results from DLS measurements of the particles' size distribution by number and by intensity for PF-PEG350, PF-PEG2000 and PF-PEG5000 Pdots. Distribution by numbers is preferred, because the presentation of distribution by intensity gives a much higher weight to very large particles, even if they consist a small minority of the total population,25 as seen in Fig. S1.† The distributions showed two main size populations in each case. For PF-PEG350 Pdots, nearly 98% of particles have sizes in the range 30.9–124 nm and the mean size is 46 ± 1.8 nm, whereas 2% have mean size of 307.4 ± 53 nm. For PF-PEG2000 Pdots, 52% of particles have sizes in the range 62–158 nm and the mean size is 107 ± 8 nm, whereas 48% have mean size of 399 ± 1 nm and for PF-PEG5000 Pdots, 98% of particles have sizes in the range 48–91 nm and the mean size is 75 ± 4 nm, whereas 2% have mean size of 378 ± 37 nm. One should notice that if the PEG chains were fully stretched the difference between the diameters would have been bigger than those observed here. This indicates that the chains form a thick layer of PEG chains wrapping the core of the Pdots.
The morphology and size of the Pdots can be also characterized by Atomic Force Microscopy. Fig. S2† shows images of PF-PEG350, PF-PEG2000 and PF-PEG5000. The closely spherical shape of these Pdots can be seen in the figure. The average diameter for PF-PEG350, PF-PEG2000 and PF-PEG5000 are 43 ± 9, 77 ± 0.3 and 60 ± 0.15 nm, respectively.
The fluorescence quantum yields of the Pdots were determined by reference to rhodamine 590, whose yield in ethanol is 0.94.26 The calculated fluorescence quantum yields were 0.31, 0.30 and 0.29 for PF-PEG350, PF-PEG2000 and PF-PEG5000, respectively.
FRET efficiency
We measured the efficiency of FRET,27–31 E, by monitoring the donor's (Pdot) steady-state emission intensity, I, as well as its emission decay time, τ, when alone and when in the presence of the RB or RBME sensitizers, serving as acceptors, using eqn (3).
 | (3) |
| R06 = 8.79 × 10−5κ2n−4Q0J | (4) |
| J = ∫fD(λ)εA(λ)λ4dλ | (5) |
We calculated for each donor–acceptor pair, the overlap integral which is shown in Fig. 1c, using eqn (5). For example, for the PF-PEG350 Pdot–RB dyad (upper panel) it is 8.55 × 10−13 M−1 cm3, while for the same Pdot with RBME (lower panel) it amounts to 3.26 × 10−13 M−1 cm3. The R0 that we obtained were 6.06 nm and 5.16 nm, respectively. These relatively high values,34 which are a consequence of the good spectral overlap as can be seen in Fig. 1c, enable efficient resonance energy transfer even at higher donor–acceptor distances.
The analysis of fluorescence time-resolved FRET experiments is based on curve fitting methods. These methods yield the theoretical decay curve best fitted to the experimentally recorded fluorescence decay curves. We obtained the average fluorescence life-time, 〈τ〉, of Pdots with different shell compositions. The average life-time of the samples was determined from multiexponential analysis of the emission decay:  , where ai and τi are the relative amplitude and the life-time of the ith decay component respectively (Table 1). The quality of the fits of the experimental and calculated curves is judged by the minimization to the χ2 values.
, where ai and τi are the relative amplitude and the life-time of the ith decay component respectively (Table 1). The quality of the fits of the experimental and calculated curves is judged by the minimization to the χ2 values.
| Sample | τ1 (a1) | τ2 (a2) | τ3 (a3) | 〈τ〉 (ns) | χ2 |
|---|---|---|---|---|---|
| PF-PEG350 | 1.56 (0.31) | 0.74 (0.49) | 0.16 (0.2) | 0.88 | 1.15 |
| PF-PEG2000 | 2.10 (0.09) | 0.51 (0.49) | 0.14 (0.41) | 0.51 | 0.97 |
| PF-PEG5000 | 1.25 (0.36) | 0.55 (0.42) | 0.08 (0.22) | 0.71 | 1.01 |
In the presence of increasing concentrations of the acceptor RB or RBME, a drastic shortening of the decay time is observed. We show the time-resolved experiment for the PF-PEG350 Pdot with increasing concentrations of RB (see Fig. S3†). The average decay time of the Pdot reduces from 0.88 ns to 0.17 ns at higher concentration of RB. This fact confirms the energy transfer process from PF Pdot to RB. All average decay times of PF Pdots with different shell compositions upon addition of RB or RBME due to time-resolved measurements are shown in Fig. 2.
 | ||
| Fig. 2 Change in the averaged fluorescence life-time decay of Pdots with different shell compositions upon addition of Rose Bengal (RB) or Rose Bengal Methyl Ester (RBME). (a) 4.1 × 10−3 mg mL−1 PF-PEG350, (b) 2.1 × 10−4 mg mL−1 PF-PEG2000 and (d) 1.1 × 10−3 mg mL−1 PF-PEG5000 Pdots (λex = 445 nm, λem = 531 nm). The curves depict the trend of the fluorescence life-time decrease. | ||
As can see, FRET is efficient even at low concentration as 0.1 μM of RB or RBME. Based on the fluorescence life-time measurements, as shown in Fig. 2, and using eqn (3), the FRET efficiencies for the Pdot–RB and Pdot–RBME dyads were calculated, and the results for the PF-PEG350 Pdots are shown in Fig. 3.
 | ||
Fig. 3 Efficiency of fluorescence resonance energy transfer. The efficiency curves depict the trend of the increase in energy transfer from 1.2 × 10−4 mg mL−1 PF-PEG350 Pdots to Rose Bengal, employing the donor's fluorescence intensity ( ) and from 4.1 × 10−3 mg mL−1 PF-PEG350 Pdot to Rose Bengal ( ) and from 4.1 × 10−3 mg mL−1 PF-PEG350 Pdot to Rose Bengal ( ) and to Rose Bengal Methyl Ester ( ) and to Rose Bengal Methyl Ester ( ) obtained from the donor's fluorescence decay times. ) obtained from the donor's fluorescence decay times. | ||
For steady state measurements of FRET, the fluorescence spectra of the PF-Pdots in aqueous suspensions were measured while the RB photosensitizer was added at increasing concentrations. We show as an example, in Fig. 1d, the emission spectra of PF-PEG350 Pdots, upon titration with increasing concentrations of RB. The Pdots' fluorescence intensity is seen to decrease and, simultaneously, RB's fluorescence intensity increases, as a consequence of FRET. The absorbance of RB at the excitation wavelength that we used is minimal, thus no direct excitation of RB was achieved under these conditions and no fluorescence intensity was discovered. The fluorescence spectra were resolved into two emission bands with Lorentzian shapes, and the calculated FRET efficiencies are shown in Fig. 3.
Very high energy transfer efficiencies (>60%) are obtained even at a 1 μM concentration of the acceptor, as evident from the donor's steady state fluorescence intensities as well as from its fluorescence decay time. This is explainable on the good spectral overlap that is necessary for FRET, which is manifested in the relatively high R0. It is born out in practice probably by the close contact between donor and acceptor, because the latter is brought to close proximity by its intercalation into the thin enveloping PEGylated layer around the Pdots. This also explains the higher FRET efficiency that is obtained with the PEG350 coating (Fig. 2a). The lipophilic coating environment is thinner than the other PEG's length and the sensitizer that is intercalated into it is located closer to the core of the polymeric dye.
Relative singlet oxygen quantum yields
To measure the production yield of singlet oxygen by direct excitation of the photosensitizer or via FRET from the Pdots, we employed DMA as a chemical target that is peroxidized by singlet oxygen, whereby its fluorescence is diminished. We measured, in a time-drive mode, the kinetics of the decrease of DMA's fluorescence intensity with the presence of PF Pdots, which consist of different shell compositions and when they are with the acceptor RB or RBME (Fig. S4†), while a 473 nm laser beam irradiates the sample and drives the photochemical process. The fluorescence intensity decay was fitted to an exponential equation:| IDMA = Ae−kDMAt | (6) |
 | (7) |
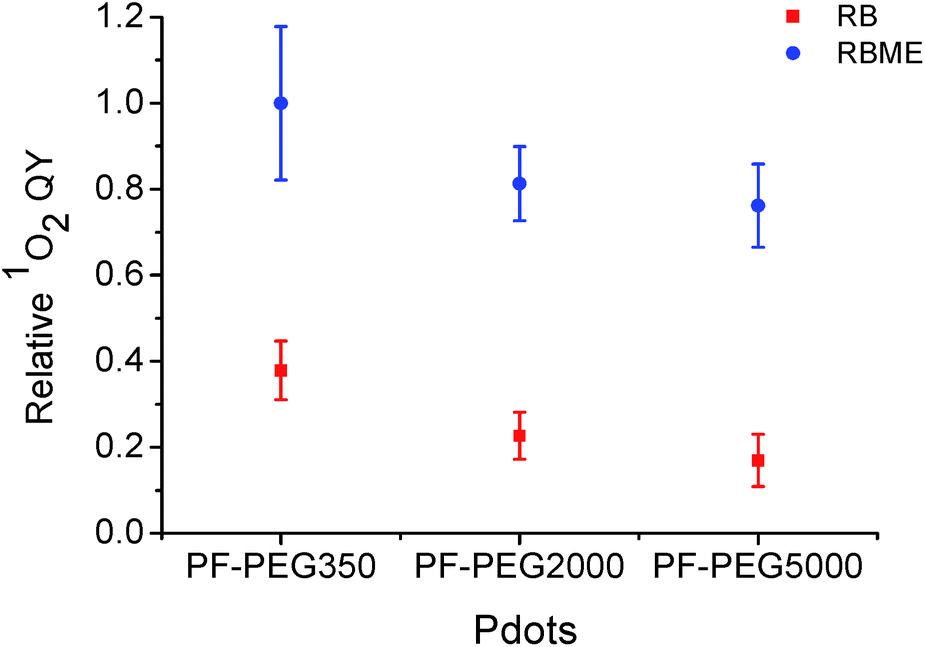 | ||
| Fig. 4 Relative singlet oxygen quantum yields. Enhancement of singlet oxygen production by Pdots-RBME dyad compare to Pdots–RB dyads (±calculated from at least three independent measurements). | ||
As can be seen in Fig. 4, the generation of singlet oxygen of Pdots–RBME is significantly increased, compared to that of Pdots–RB. The data in the figure also indicate that the best singlet oxygen production via FRET is achieved, for both sensitizers, with Pdots of PF-PEG350. We attribute this to the thinner envelope of PEGylated lipids that surround the core of the Pdot, which brings the sensitizer to a closer proximity to the organic chromophoric polymer, thus enhancing FRET. RBME being more hydrophobic than RB, partitions deeper into the PEGylated shell of the Pdot and locate closer to the Pdot's core, which leads to more efficient FRET between them.
We chose to continue the cellular studies with RB, being reasonably water-soluble so that aggregation phenomena will be avoided in cells' suspensions.
Uptake of Pdots by MCF-7 cells
Following the examination of basic spectroscopic properties of the Pdot-sensitizer dyads and the energy transfer process between them in aqueous solutions, we studied the uptake of the dyads by MCF-7 breast cancer cells. The uptake is naturally a prerequisite for their photobiological activity. Our purpose was to examine the extent of cell penetration by these particles and to probe the photodamage to cells as a result of photosensitization, which occurred by FRET. Fig. 5 shows white-light and fluorescence microscopy pictures of PF-PEG350 and PF-PEG2000 Pdots. | ||
| Fig. 5 Fluorescence and white-light composed micrographs of Pdots taken up by MCF-7 cancer cells. (a) Control cells (b) cells with PF-PEG350 Pdots (c) cells with PF-PEG2000 Pdots. The cells were incubated with the Pdots for 4 hours, at 37 °C. | ||
The PF-PEG350 Pdots enter the cells much better than PF-PEG2000 Pdots, and are distributed in the intracellular cytoplasm. The reason for the difference between the extent of uptake of the two types of Pdots could arise either from the PEG2000 forming larger, bulkier, particles or from the more PEGylated coating being endowed with higher water solubility.
For a quantitative assessment, MCF-7 cells were incubated with the Pdots and the intracellular fluorescence intensity was measured by flow cytometry, FACS. We show in Fig. 6a, the uptake of PF-PEG350 Pdots by the cells, as a function of the duration of incubation, at 37 °C and 4 °C. The comparison of uptake at these two temperatures points to the strong involvement of endocytosis, rather than passive diffusion, in the uptake of these nanoparticles by the cells. Endocytosis is an efficient process at 37 °C and is drastically slowed down at 4 °C, as is seen to occur here, with minimal increase in the cellular fluorescence intensity, even after 3 hours of incubation.
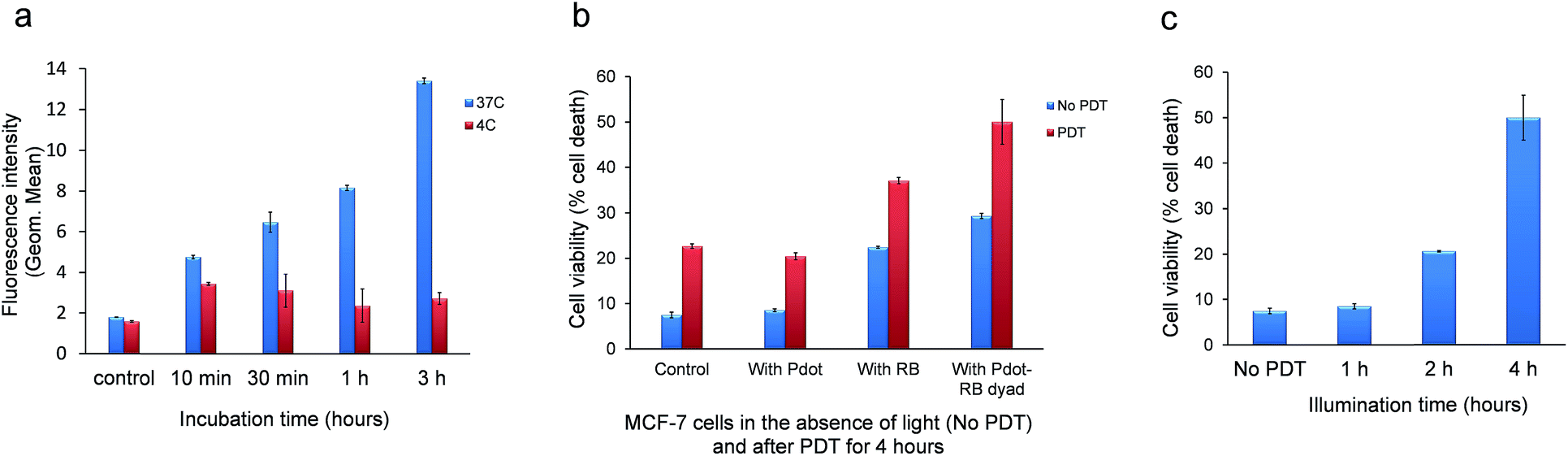 | ||
| Fig. 6 Pdots' penetration into cells and the cells' damage caused by illumination (473 nm, 0.0015 J cm−2) in the presence of Pdots, RB and Pdot–RB dyad. (a) Intracellular Pdots fluorescence intensity upon incubation of cells with PF-PEG350 Pdots in serum-free DMEM medium, at 37 °C (blue) and 4 °C (red). (b) Normalized integrated FACS fluorescence intensity of propidium iodide as an indicator of damage to MCF-7 cells by PF-PEG350 Pdots particles, by RB and by Pdot–RB dyad in the absence of light. (c) The progress of damage by the dyads after 1,2 and 4 hours of illumination and (d) damage after 4 hours of illumination at 473 nm. | ||
Cell viability under photosensitization with Pdot-sensitizer dyads
The principal aim of this study was the evaluation of the photodynamic damage to cancer cells that can be achieved by FRET-assisted photosensitization.Panel (b) of Fig. 6 shows the photodamage that is inflicted to cells, following photosensitization with RB alone or with PF-PEG350 Pdots or Pdots–RB dyads. The damage was measured by uptake of the viability stain propidium iodide, using FACS cytometry. Fig. 6b shows the viability of the cells under dark conditions (without illumination, blue) and it indicates that no noticeable toxicity was caused by either RB alone, by Pdots alone, or by their dyads compared to the same samples while were illuminated in a 473 nm for up to 4 hours (red). See also Fig. S5,† the histograms show samples without illumination (left side) compared to samples that involve illumination for 4 hours (right side). One can see a noticeable damage that was inflicted by the dyad only (Fig. S5d,† right side), where the peaks (PI fluorescence) in the histogram are different in their height and location than the peaks in the histograms in the left side. In Fig. S5d† with illumination, the averaged fluorescence intensity of the left peak is 103 which is 10 times greater of PI fluorescence than in other histograms. This evidence shows that more cells are damaged and started their apoptosis. Fig. 6c shows the progress of damage to the cells as the illumination is prolonged (see also Fig. S6†). This figure shows the enhancement in the photodamage effect when FRET is involved in transducing light energy from the Pdot to the sensitizer. For estimating the cell damage we use the gating on histogram of cells stained with PI and the appeared percentage represent the dead cells population. As can be seen in this figure, as the illumination is prolonged the percentage of the dead cells population is higher. Almost 6 times higher for 4 hours illumination compared to 1 hour of illumination at the same conditions, in average 50% compared to 8.5% respectively. To induce FRET from Pdots to RB molecules the intracellular fluorescence was measured using an ImageStream, a high-resolution imaging flow cytometer. As can see in Fig. 7, we used laser lines at 488 nm, to excite Pdots, and 561 nm, to excite RB. The fluorescence was collected in CH2, emission bandwidth of Pdot's fluorescence and CH3, emission bandwidth of RB's fluorescence. Cells containing Pdots (ex: 488 nm) have a strong fluorescence at wavelengths of 480–560 nm (CH2) and a weak fluorescence at wavelengths of 560–595 nm (CH3). But when cells contain Pdot–RB dyads (ex: 488 nm) there is a strong emission at CH3 and FRET is induced compared to cells contain Pdot–RB dyad (ex: 561 nm). The results shown in this figure were quantified automatically by the ImageStream flow cytometer over the whole population of thousands of cells. The data of the mean fluorescence intensities is seen in Table S1.† The effect of FRET is seen in the boldface value in the table.
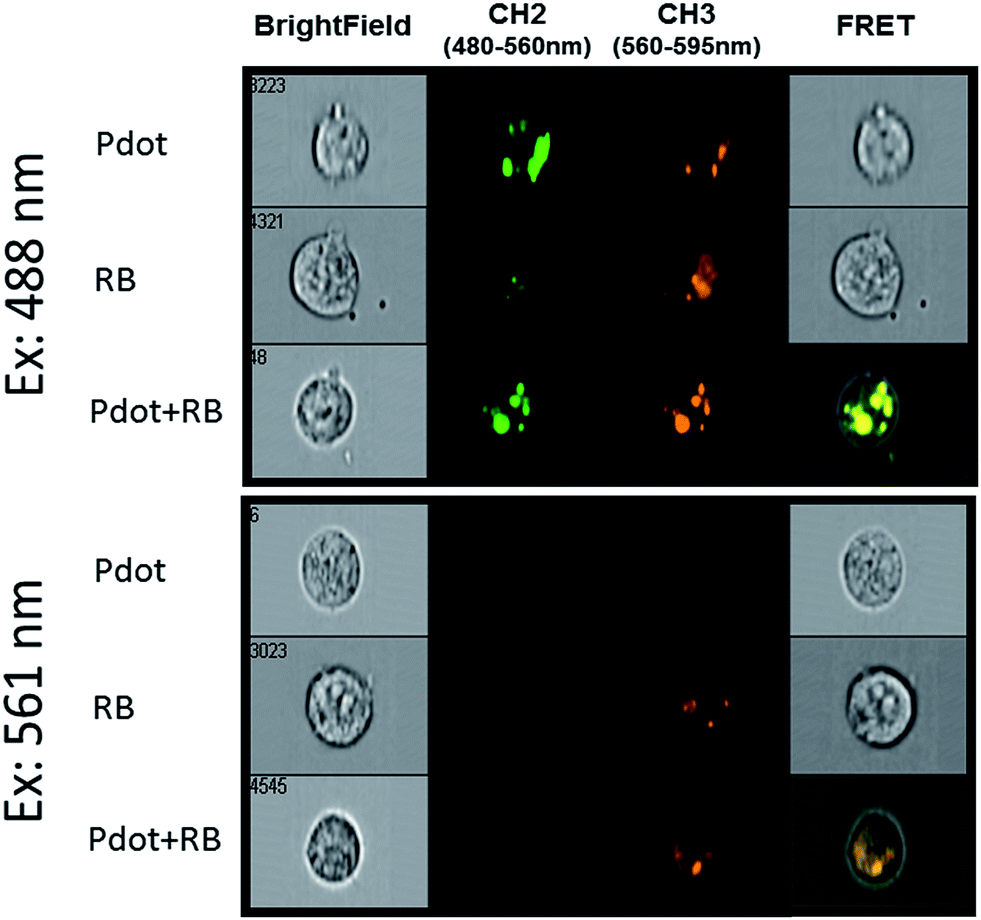 | ||
| Fig. 7 Cellular fluorescence of MCF-7 cells with PF-PEG350 Pdots, with RB and with Pdot–RB dyad. Each cell is represented by a row of four images. From left to right: bright field, CH2 (green fluorescence 480–560 nm which represents the Pdot's emission), CH3 (orange fluorescence 560–595 nm which represents the RB's emission) and FRET (represented by the co-localization) with the two excitation wavelengths shown on the left. | ||
To evaluate the stability of the dyads, fluorescence spectra of the Pdot–RB complex in biological DMEM media containing serum were measured for 4 hours, which is the longest duration of the process of photosensitization. These spectra showed a good stability without any release of the RB from the Pdot–RB dyad complex. In addition, for demonstrating the stability of the materials in cells, they were incubated with cells for 24 hours and measured by the ImageStream flow cytometer. In Fig. S6† we show the cellular fluorescence which is captured by CH2 and CH3 when the excitation was at 488 nm. They show good emission of both materials in the cells, which indicates the presence of the dyads and the induced FRET, which is represented by the co-localization (seen in the right column), even after this long incubation in cells. The E-SEM pictures in Fig. 8 show representative cells that were treated with PF-PEG350 Pdot and Rose Bengal and which were illuminated for 1, 2 and 4 hours. As can seen, prolonging the illumination time increases the damage that is caused to the cells.
 | ||
| Fig. 8 E-SEM pictures of MCF-7 cells. (a) Cells without treatment, which represent the control cells and cells with PF-PEG350 Pdot–RB dyads after PDT treatment of (b) 1 hour, (c) 2 hours and (d) 4 hours. | ||
Conclusions
Pdot nanoparticles are prepared from semiconducting π-conjugated chromophoric polymers, and by mixing them with PEGylated phospholipids they can be made water dispersible, forming stable colloids in aqueous media. The PEGylated moieties that cover the Pdot form a nano-phase into which amphiphilic molecules can partition. These aqueous Pdot solutions are very stable for months and exhibit a very good fluorescence and absorbance. We show in this paper that the photosensitizers Rose Bengal and Rose Bengal Methyl Ester partition very efficiently into the coating of the Pdots. The close contact and the good overlap between the Pdots' emission band and RB's absorption band facilitate efficient energy transfer from the Pdots to the photosensitizers. The production of singlet oxygen, the active oxygen species that is involved in the photodynamic action, is enhanced in a homogeneous aqueous solution. It is also enhanced in the process of photodynamic damaging of MCF-7 cells, which take up the Pdots by an endocytotic mechanism. Therefore, the convenient, non-covalent attachment of photosensitizer molecules to Pdots enables a more efficient use of the light's bandwidth, since light can be used not only at the usually red absorption band of the photosensitizer, but rather the whole spectral range that is covered by the broad absorption band of the Pdots becomes available for the photobiological process by employing a non-coherent, broad-band light source. Indeed, shorter wavelengths will penetrate less into live tissue, but one can only gain from absorption of light by the chromophoric polymers, that have a higher optical cross-section, and that will transfer energy, by FRET, to the sensitizer. Surely, despite the fact that the QY of the photosensitizers is almost maximal, the possibility to transduce energy from the Pdots is greater due to the integrate cross-section of the Pdots. The absorption by these Pdots can even be carried out by broad-band, non-coherent light sources. At the same time, direct illumination of the sensitizer which is in the dyad can be achieved in parallel with the FRET mechanism, as is done by a usual photosensitization protocol. Thus, the two pathways can function in parallel. This study demonstrates that Pdots have a bright prospect not only in cellular labeling, imaging, tracking and sensing,35 but also in the field of biological photosensitization. Undoubtedly, further research will result in major improvements in this technique. When the technique has been fully developed and evaluated, it will surely play an important role in the management of photodynamic therapy.Acknowledgements
We thank Prof. Elisha Haas and Dr Dan Amir, from Bar Ilan University for the measurements of the fluorescence decays. This research was supported by the Michael David Falk Chair in Laser Phototherapy.Notes and references
- J. K. Jaiswal and S. M. Simon, Potentials and Pitfalls of Fluorescent Quantum Dots for Biological Imaging, Trends Cell Biol., 2004, 14, 497–504 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Applications of Nanoparticles in Biology, Adv. Mater., 2008, 20, 4225 CrossRef CAS.
- Nanotechnology in Biology and Medicine, Methods, Devices, and Applications, ed. T. Vo-Dinh, CRC Press, Boca Raton, FL, 2007 Search PubMed.
- K. K. Jain, ed. The Handbook of Nanomedicine, Humana Press, N.Y., 2nd edn, 2012 Search PubMed.
- C. R. L. Lipson, E. J. Baldes and A. M. Olsen, The Use of a Derivative of Hematoporphyrin in Tumor Detection, J. Natl. Cancer Inst., 1961, 26, 1–8 Search PubMed.
- T. J. Dougherty, J. E. Kaufman, A. Goldfarb, K. R. Weishaupt, D. Boyle and A. Mittleman, Photoradiation Therapy for the Treatment of Malignant Tumors, Cancer Res., 1978, 38, 2628–2635 CAS.
- T. J. Dougherty, Photosensitizers: Therapy and Detection of Malignant Tumors, Photochem. Photobiol., 1987, 45, 879–889 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, Photodynamic Therapy, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon & Breach Science, Amsterdam, 2000 Search PubMed.
- M. Wainwright, Photoinactivation of Viruses, Photochem. Photobiol. Sci., 2004, 3, 406–411 CAS.
- A. E. O'Connor, W. M. Gallagher and A. T. Byrne, Porphyrin and Nonporphyrin Photosensitizers in Oncology: Preclinical and Clinical Advances in Photodynamic Therapy, Photochem. Photobiol., 2009, 85, 1053–1074 CrossRef PubMed.
- M. Ethirajan, Y. H. Chen, P. Joshi and R. K. Pandey, The Role of Porphyrin Chemistry in Tumor Imaging and Photodynamic Therapy, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- C. Szymanski, C. F. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, Single Molecule Nanoparticles of the Conjugated Polymer MEH-PPV, Preparation and Characterization by Near-Field Scanning Optical Microscopy, J. Phys. Chem. B, 2005, 109, 8543–8546 CrossRef CAS PubMed.
- C. F. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, Multicolor Conjugated Polymer Dots for Biological Fluorescence Imaging, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, Phospholipid Encapsulated Semiconducting Polymer Nanoparticles: Their Use in Cell Imaging and Protein Attachment, J. Am. Chem. Soc., 2010, 132, 3989–3996 CrossRef CAS PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, Bioconjugation of Ultrabright Semiconducting Polymer Dots for Specific Cellular Targeting, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- S. Bhattacharyya, M. K. Barman, A. Baidya and A. Patra, Singlet Oxygen Generation from Polymer Nanoparticles–Photosensitizer Conjugates Using FRET Cascade, J. Phys. Chem. C, 2014, 118, 9733–9740 CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, Rate Constants For the Decay and Reactions of the Lowest Electronically Excited Singlet State of Molecular Oxygen in Solution. An Expanded and Revised Compilation, J. Phys. Chem. Ref. Data, 1995, 24, 663–1021 CrossRef CAS PubMed.
- P. R. Bevington, Data Reduction and Error Analysis for the Physical Sciences, McGraw-Hill, New York, USA, 1969, ch. 5 Search PubMed.
- H. Gamliel, Optimum Fixation Conditions May Allow Air Drying of Soft Biological Specimens with Minimum Cell Shrinkage and Maximum Preservation of Surface-Feature, Scanning Electron Microsc., 1985, 4, 1649–1664 Search PubMed.
- J. M. Tsay, M. Trzoss, L. Shi, X. Kong, M. Selke, M. E. Jung and S. Weiss, Singlet Oxygen Production by Peptide-Coated Quantum Dot–Photosensitizer Conjugates, J. Am. Chem. Soc., 2007, 129, 6865–6871 CrossRef CAS PubMed.
- J. L. Grimland, C. Wu, R. R. Ramoutar, J. L. Brumaghim and J. McNeill, Photosensitizer-Doped Conjugated Polymer Nanoparticles with High Cross-Sections for One- and Two-Photon Excitation, Nanoscale, 2011, 3, 1451–1455 RSC.
- D. C. Neckers, Rose Bengal, J. Photochem. Photobiol., A, 1989, 47, 1–29 CrossRef CAS.
- M. Schafer, C. Schmitz, R. Facius, G. Horneck, B. Milow, K. H. Funken and J. Ortner, Systematic Study of Parameters Influencing the Action of Rose Bengal with Visible Light on Bacterial Cells: Comparison Between the Biological Effect and Singlet-Oxygen Production, Photochem. Photobiol., 2000, 71, 514–523 CrossRef CAS.
- R. Calderon-Villajos, C. Zaldo and C. Cascale, Micro- and Nanosized Architectures in Hydrothermal Tm3+-Doped GdVO4: Chemical Insights Towards Preservation of the Emission Efficiency, CrystEngComm, 2012, 14, 2756–2768 RSC.
- M. Fischer and J. Georges, Fluorescence Quantum Yield of Rhodamine 6G in Ethanol as a Function of Concentration Using Thermal Lens Spectrometry, Chem. Phys. Lett., 1996, 260, 115–118 CrossRef CAS.
- E. Haas, The Study of Protein Folding and Dynamics by Determination of Intramolecular Distance Distributions and their Fluctuations Using Ensemble and Single-Molecule FRET Measurements, ChemPhysChem, 2005, 6, 858–870 CrossRef CAS PubMed.
- L. E. Fisher, D. M. Engelman and J. N. Sturgis, Detergents Modulate Dimerization, But Not Helicity of the Glycophorin a Transmembrane Domain, J. Mol. Biol., 1999, 293, 639–651 CrossRef CAS PubMed.
- R. M. Johnson, C. L. Heslop and C. M. Deber, Hydrophobic Helical Hairpins: Design and Packing Interactions in Membrane Environments, Biochemistry, 2004, 43, 14361–14369 CrossRef CAS PubMed.
- M. You, E. Li, W. C. Wimley and K. Hristova, Forster Resonance Energy Transfer in Liposomes: Measurements of Transmembrane Helix Dimerization in the Native Bilayer Environment. Anal, Biochemistry, 2005, 340, 154–164 CAS.
- B. Corry, P. Rigby, Z. W. Liu and B. Martinac, Conformational Changes Involved in MscL Channel Gating Measured Using FRET Spectroscopy, Biophys. J. Lett., 2005, 89, L49–L51 CrossRef CAS PubMed.
- J. R. Lakowicz, Energy Transfer, in Principles of Fluorescence Spectroscopy, Springer Science & Business Media, New York, 13th edn, 2006, pp. 443–476 Search PubMed.
- Modern Quantum Chemistry, ed. O. Sinanoglu, Academic Press, New York, 1965, vol. III, pp. 93–137 Search PubMed.
- B. W. Van der Meer, G. Coker III and S. Y. Simon Chen, Resonance Energy Transfer: Theory and Data, VCH, New York, 1994 Search PubMed.
- C. Wu and D. T. Chiu, Highly Fluorescent Semiconducting Polymer Dots for Biology and Medicine, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15291c |
| This journal is © The Royal Society of Chemistry 2015 |