
Spectroscopic characterization of C-4 substituted 3,5-dichloro-4H-1,2,6-thiadiazines†
Abstract
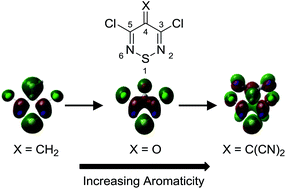
Three 3,5-dichloro-4H-1,2,6-thiadiazines, which differ according to the electron withdrawing nature of their substituent at C-4: (a) 3,5-dichloro-4-methylene-4H-1,2,6-thiadiazine (5a), (b) 3,5-dichloro-4H-1,2,6-thiadiazin-4-one (5b) and (c) 2-(3,5-dichloro-4H-1,2,6-thiadiazin-4-ylidene)malononitrile (5c), are characterized using resonance Raman (RR), absorption (UV/vis) and photoluminescence (PL) spectroscopies. These weakly aromatic and electron-deficient heterocycles are potential components as acceptors in donor–acceptor systems for organic electronics. Experimental results, which include the synthesis, characterization and single crystal X-ray structure of 3,5-dichloro-4-methylene-4H-1,2,6-thiadiazine (5a), combined with theoretical calculations of their orbitals and vibrational frequencies, provide an understanding of the optical properties, on the basis of molecular geometry and electron distribution.
 Please wait while we load your content...
Please wait while we load your content...

