
Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES): as a new, green and efficient nanocatalyst for preparation of 5-substituted-1H-tetrazoles†
Nasrin Razavi and
Batool Akhlaghinia*
Department of Chemistry, Faculty of Sciences, Ferdowsi University of Mashhad, Mashhad 9177948974, Iran. E-mail: akhlaghinia@um.ac.ir; Fax: +98-51-3879-5457; Tel: +98-51-3880-5527
First published on 14th January 2015
Abstract
Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES) is a novel and efficient heterogeneous nanocatalyst in the [3 + 2] cycloaddition reactions of various organic nitriles with sodium azide. The protocol can provide a series of 5-substituted-1H-tetrazoles under mild conditions in DMSO. Efficient transformation, mild reaction conditions, easy product isolation and the potential reusability of the catalyst are attractive features. The catalyst (CAES) was characterized by FT-IR, TGA, TEM, BET, SEM-EDS, CHN and ICP techniques.
Introduction
Recently, catalytic synthesis of tetrazoles which represent an important class of heterocycles, has acquired immense attention, due to their wide ranging applications. Tetrazoles are used in materials as specialty explosives and information recording systems, in pharmaceuticals as lipophilic spacers and carboxylic acid surrogates, in coordination chemistry as ligands and also used as precursors in a variety of nitrogen containing heterocycles.1 The proton acid-catalyzed cycloaddition between hydrazoic acid and nitriles has long been one of the main routes to 5-substituted-1H-tetrazoles. This method suffers from explosion of a large amounts of harmful hydrazoic acid.2 As, it is necessary to overcome the above mentioned drawback and to improve the synthetic methods of obtaining 5-substituted-1H-tetrazoles, a number of new catalysts have also been investigated till date. Most of them which are based on [3 + 2] cycloaddition reaction between azide ion and organic nitriles are imidazole-based zwitterionic-type molten salts,3 β-cyclodextrin,4 copper triflates,5 CdCl2,6 Fe(OAc)2,7 Zn(II) salts,8–10 AgNO3,11 Et3N·HCl,12 Lewis acids such as B(C6F5)3,13 AlCl3,14 BF3·OEt2,15 FeCl3,16 TBAF,17 InCl3,18 I2,19 (CH3)2SnO,20 and Bronsted acid catalyst.21 Also, the reactions were carried out by using some heterogeneous catalysts such as chitosan derived magnetic ionic liquid,22 tungstates,23 NaHSO4·SiO2,19 FeCl3–SiO2,16 COY zeolites,24 mesoporous ZnS nanospheres,25 Zn/Al hydrotalcite,26 nanocrystalline ZnO,27 ZnO/Co3O4,28 Zn–Cu alloy,29 Zn hydroxyapatite,30 Pd(PPh3)4,31 Cu2O,32 AgNPs,33 WAlPO-5 microspheres,34 Fe3O4@SiO2/salen of Cu(II)35 and CuFe2O4 nano particles.36Unfortunately, with very few exceptions, earlier synthetic methods suffered from various disadvantages including the use of expensive and toxic metals, expensive reagents, strong Lewis acids, long reaction times (several hours to days) in combination with high reaction temperatures, low yield, harsh reaction conditions, tedious work-ups and difficulty in the separation and the recovery of the catalyst. In addition to this in some cases, formation of highly volatile and toxic hydrazoic acid as by product warrant the safety of the method.37 As, the increasing demands of environmental legislation promotes the chemical industries to minimize waste production in chemical manufacture,38 a growing interest has been witnessed in the use of heterogeneous catalyst, in different areas of organic synthesis. Organic–inorganic hybrid catalysts, such as organosilane39 having a covalently anchored organic spacer as a class of tethered heterogeneous catalysts have been widely applied due to their environmental compatibility combined with good yield, selectivity and easy separation.40
In the present study, we would like to report a new silica-supported catalyst based on Cu(II) immobilized on aminated epichlorohydrin. Designing and the application of this heterogeneous, more convenient and reusable catalyst in the [3 + 2] cycloaddition reactions of various organic nitriles with sodium azide were investigated. At first, the silica surface was activated by refluxing hydrochloric acid solution (37%) to increase the terminal silanol–OH groups (I).41 Then the support was heated at reflux with an aqueous solution of NaOH (0.04 M) to change the terminal silanol–OH groups into negatively charged silanol–O− groups to produce the desired activated silica (II). The activated silica was suspended to pure epichlorohydrin at 60 °C with vigorous stirring and then the resulting epoxy activated silica (III) reacted with ammonia solution. The aminated epichlorohydrin activated silica (IV) was used as the support and ligand for entrapment of Cu(II) by the reaction of (IV) with a solution of Cu(OAc)2·H2O. The resulting complex contains Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES) (V) (Scheme 1).
 | ||
| Scheme 1 Preparation of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
Results and discussion
After preparation and characterization of CAES and in continuation of our recent studies on the application of CuSO4·5H2O,42,43 in organic reactions, the catalytic activity of CAES as a new nanocatalyst was evaluated in [3 + 2] cycloaddition reactions of various organic nitriles with sodium azide to produce a library of 5-substituted-1H-tetrazoles (Scheme 2).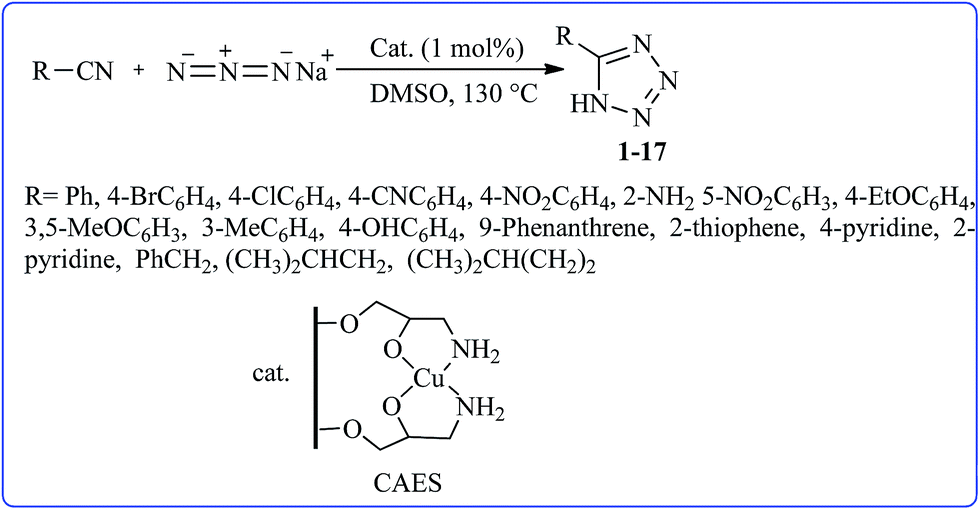 | ||
| Scheme 2 Synthesis of different structurally 5-substituted-1H-tetrazoles in the presence of CAES in DMSO. | ||
To obtain the best conditions for the synthesis of titled compounds, the reaction of benzonitrile and sodium azide was selected as a model reaction and was studied in the presence of CAES by applying different molar ratios of reactants as well as solvents at different temperatures. The obtained results are summarized in Table 1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaN3 and different temperatures
:NaN3 and different temperatures
| Entry | Molar ratio (nitrile:NaN3) |
Catalyst (g) | Solvent | Temperature (°C) | Time (h) | Isolated yield (%) |
|---|---|---|---|---|---|---|
| a The reaction was performed in the presence of IV.b The reaction was performed in the presence of Cu(OAC)2·H2O. | ||||||
| 1 | 1:1 |
— | DMSO | 130 | 24 | 35 |
| 2a | 1:1 |
0.04 | DMSO | 130 | 24 | 35 |
| 3 | 1:1 |
0.04 (1 mol%) | DMSO | 130 | 1 | 95 |
| 4 | 1:1 |
0.04 (1 mol%) | DMSO | 120 | 1 | 70 |
| 5 | 1:1 |
0.04 (1 mol%) | DMSO | 80 | 4 | 50 |
| 6 | 1:1 |
0.04 (1 mol%) | DMSO | 140 | 1 | 95 |
| 7 | 1:1 |
0.08 (2 mol%) | DMSO | 140 | 1 | 95 |
| 8 | 1:1 |
0.04 (1 mol%) | — | 130 | 1 | 20 |
| 9 | 1:1 |
0.04 (1 mol%) | DMF | 130 | 1 | 60 |
| 10 | 1:1 |
0.04 (1 mol%) | CH3CN | 130 | 1 | 20 |
| 11 | 1:1 |
0.04 (1 mol%) | H2O | 130 | 1 | Trace |
| 12 | 1:1 |
0.04 (1 mol%) | 1,4-Dioxane | 130 | 1 | 5 |
| 13 | 1:1.5 |
0.04 (1 mol%) | DMSO | 130 | 1 | 95 |
| 14b | 1:1 |
0.0018 (0.9 mol%) | DMSO | 130 | 1 | 50 |
At first we used DMSO as solvent for the reaction between 1:1 molar ratio of benzonitrile:sodium azide at 130 °C in the absence of any catalyst. After 24 h, 5-phenyl-1H-tetrazole was afforded in 35% isolated yield (Table 1, entry 1). The same result was obtained when the reaction was performed in the presence of aminated epichlorohydrin activated silica (IV) (Table 1, entry 2). This means that IV has not any catalytic activity in [3 + 2] cycloaddition. Applying Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES) (0.0400 g, 1 mol% contains 0.0006 g, 0.01 mmol of Cu) as catalyst in cycloaddition reaction produced the desired product in high yield after 1 h (Table 1, entry 3). The presence of CAES has great influence on reaction time as well as yield (Table 1, entry 3). To investigate the effect of temperature on the reaction rate, [3 + 2] cycloaddition was performed at different temperatures (Table 1, entries 4–6). Lower temperatures reduced the rate of reaction considerably while higher temperature (140 °C) as well as higher catalyst loading (0.0800 g, 2 mol% contains 0.0012 g, 0.02 mmol of Cu, entry 7) has not any influence on the reaction rate. In an effort to develop better reaction conditions, solvent free condition and different solvents were screened for the preparation of 5-phenyl-1H-tetrazole in the presence of 0.0400 g, 1 mol% of CAES at 130 °C (Table 1, entries 8–12). As can be seen, the catalytic activity of CAES was decreased efficiently in solvent free condition as well as DMF, CH3CN, H2O and 1,4-dioxane. Further increase in reaction time has no significant effect on yields. The solvent is known to significantly affect the reaction rate. The use of aqueous solvent was ineffective (entry 11), however, DMSO was found to be successful, although the superior effect of DMSO remains uncertain in comparison with the other polar solvents. To improve the yield of reaction, the effect of different molar ratios of reactants was examined (Table 1, entry 13). According to this study, the increase in amount of NaN3 from 1 to 1.5 mmol was found to have no effect on the yield. In order to ascertain the major role of CAES as a catalyst in the [3 + 2] cycloaddition, the model reaction was studied in the presence of the same amount of Cu (0.01 mmol, 0.0006 g) using 0.0018 g, 0.9 mol% of Cu(OAc)2·H2O. After appropriate reaction time (1 h), the reaction was proceeded with only 50% isolated yield and no additional conversion was obtained even after longer reaction time. Therefore, we may conclude that the catalytic activity of Cu(II) in cycloaddition reaction was improved by immobilization on aminated epichlorohydrin activated silica matrix (IV) (Table 1, entry 14).
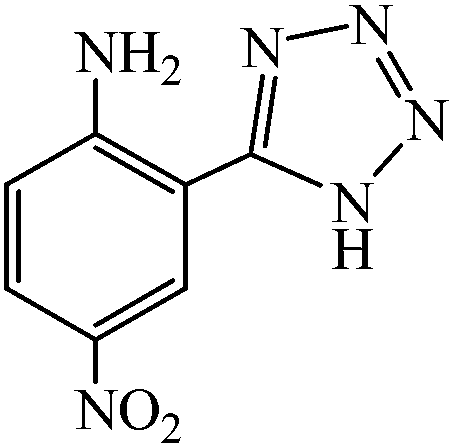
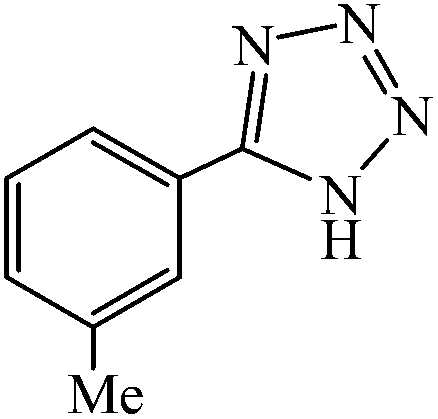
A variety of structurally divergent nitriles possessing a wide range of functional groups (Scheme 2) selected to explore the generality and scope of [3 + 2] cycloaddition reaction catalysed by CAES. The obtained results are summarized in Table 2. The optimized reaction conditions (1:1 molar ratio of nitrile:sodium azide, 0.0400 g, 1 mol% of catalyst, in DMSO at 130 °C) were used for all organic nitriles and the corresponding 5-substituted-1H-tetrazoles were prepared within 1–7 h with good to excellent yields. Aromatic nitriles containing both electron withdrawing and electron donating groups underwent the conversion smoothly. The aromatic nitriles having electron donating substituents require longer reaction time (compare entries 1–6 with entries 7–10) as donating substituent on aromatic ring decreases the electrophilic character of nitrile. Cycloaddition reactions of aliphatic nitriles proceeded more slowly than cycloaddition reactions of aromatic nitriles because of deactivation of nitriles by electron donating alkyl groups, in other words, aromatic rings can facilitate the attack of azide anion to the cyano groups and stabilize the corresponding intermediates (Table 2, entries 15–17). It is noteworthy that aryl dicyano derivatives such as terephthalonitrile also reacted smoothly with sodium azide and gave only mono adduct even by using 1:2 molar ratio of terephthalonitrile:sodium azide and 1 mol% of catalyst (Table 2, entry 4).
| Entry | Substrate | Product | Time (h) | Isolated yield (%) |
|---|---|---|---|---|
| 1 |  |
 |
1 | 95 |
| 2 |  |
 |
3 | 90 |
| 3 |  |
 |
2 | 92 |
| 4 |  |
 |
1 | 96 |
| 5 |  |
 |
1 | 90 |
| 6 |  |
 |
3 | 90 |
| 7 |  |
 |
4 | 85 |
| 8 | 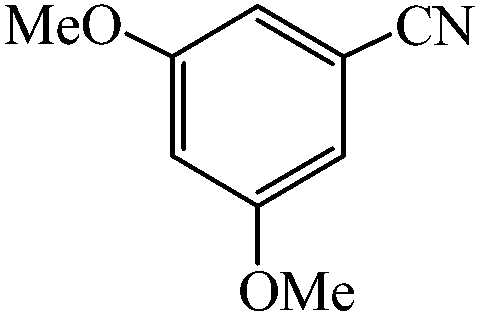 |
 |
7 | 83 |
| 9 |  |
 |
5 | 80 |
| 10 |  |
 |
7 | 80 |
| 11 |  |
 |
6 | 85 |
| 12 |  |
 |
3 | 80 |
| 13 |  |
 |
3 | 95 |
| 14 |  |
 |
5 | 80 |
| 15 |  |
 |
5 | 75 |
| 16 |  |
 |
7 | 75 |
| 17 |  |
 |
5 | 82 |
All synthesized compounds have been characterized by its melting points and FT-IR spectroscopy. The selected compounds were further identified by 1H NMR, 13C NMR spectroscopy and mass spectroscopy which compared with literature data. The disappearance of one strong and sharp absorption band (CN stretching band) and the appearance of an NH stretching band in the FT-IR spectra were evidence for the formation of 5-substituted-1H-tetrazoles. Also, the FT-IR spectra of all products show absorption bands at 1293–1233 cm−1 due to N–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, 1041–1106 cm−1 and 1189–1110 cm−1 due to tetrazole ring. In 1HNMR and 13C NMR spectra, a signal at 16.9 and 161–155 ppm are assigned to the NH and quaternary carbon NH–CN respectively.
N–, 1041–1106 cm−1 and 1189–1110 cm−1 due to tetrazole ring. In 1HNMR and 13C NMR spectra, a signal at 16.9 and 161–155 ppm are assigned to the NH and quaternary carbon NH–CN respectively.
The catalytic activity of CAES in [3 + 2] cycloaddition reactions of various organic nitriles with sodium azide is established by the fact that a long reaction time and comparatively lower yield of product was observed (Table 1, entry 1) when similar reaction was carried out in the absence of CAES. The catalytic activity of CAES could be attributed to the Lewis acidity of the Cu(II) complex. By analogy with previous reports,43 a plausible mechanism for the reaction methodology under current development is shown in Scheme 3. Initially, interaction of nitrogen atom of nitrile compound with Cu(II) forms I which accelerates the cyclization step. The [3 + 2] cycloaddition between the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of nitrile compound and azide ion takes place readily to form the intermediate II. Acidic work-up, affords III and IV. The equilibrium leads to formation of the more stable tautomer IV (5-substituted-1H-tetrazole). Nevertheless, at this time there is no experimental evidence for the [3 + 2] cycloaddition reaction proceeds in this manner, and further studies to elucidate the details of the mechanism are ongoing.
N bond of nitrile compound and azide ion takes place readily to form the intermediate II. Acidic work-up, affords III and IV. The equilibrium leads to formation of the more stable tautomer IV (5-substituted-1H-tetrazole). Nevertheless, at this time there is no experimental evidence for the [3 + 2] cycloaddition reaction proceeds in this manner, and further studies to elucidate the details of the mechanism are ongoing.
 | ||
| Scheme 3 Proposed mechanism for the synthesis of 5-substituted-1H-tetrazoles in the presence of CAES. | ||
A further advantage of this solid catalytic system is its reusability. Catalyst re-usability was assessed in the cycloaddition reaction of benzonitrile and sodium azide. To this end, the reaction was stopped after completion and the catalyst retrieved from the reaction mixture by centrifugation. For each successive use, the catalyst was washed with ethanol and acetone several times to remove the products, followed dried at 100 °C. Table 3 shows the results obtained after five re-use cycles. These results by themselves suggest that the catalyst efficiency was remained after five using. The copper content of freshly prepared CAES which was obtained by inductively coupled plasma (ICP) was 1.6092 wt%, 0.0158 g, 0.25 mmol of Cu per 1.000 g of CAES, whilst inductively coupled plasma (ICP) shows that the 5th reused CAES contains 1.2452 wt%, 0.0120 g, 0.19 mmol of Cu per 1.000 g of 5th reused CAES. It means that, in spite of some leaching of Cu(II) during recovery process of the catalyst, no significant loss of activity of the catalyst was observed and the catalytic activity maintained effectively after five reusing of CAES. In other words, the above results lead us to conclude that the amount of leached metals from the catalyst is very low.
| Entry | Run | Time (h) | Isolated yield (%) |
|---|---|---|---|
| 1 | 1 | 1 | 95 |
| 2 | 2 | 1 | 95 |
| 3 | 3 | 1 | 90 |
| 4 | 4 | 1 | 90 |
| 5 | 5 | 1 | 87 |
To show the advantage of CAES over some of the reported catalysts in the literature, the catalytic role of CAES in [3 + 2] cycloaddition reaction of benzonitrile with sodium azide was compared with various reported catalyst (Table 4). In comparison, some of these methods, in addition using environmentally hazardous catalysts and tedious purification of products, require long reaction times to achieve reasonable yields.
| Entry | Catalyst | Solvent | Temperature (°C) | Time (h) | Yield (%) | Reference |
|---|---|---|---|---|---|---|
| 1 | Chitosan derived magnetic ionic liquid | — | 70 | 7 | 87 | 22 |
| 2 | Imidazole-based zwitterionic-type molten salts | — | 120 | 12 | 84 | 3 |
| 3 | Mesoporous ZnS | DMF | 120 | 36 | 96 | 25 |
| 4 | Silica sulfuric acid | DMF | Reflux | 5 | 88 | 21c |
| 5 | AgNPs | DMF | 120 | 8 | 92 | 33 |
| 6 | Fe3O4@SiO2/Salen Cu(II) | DMF | 120 | 7 | 90 | 35 |
| 7 | B(C6F5)3 | DMF | 120 | 8 | 94 | 13 |
| 8 | AgNO3 | DMF | 120 | 5 | 83 | 11 |
| 9 | CAES | DMSO | 130 | 1 | 95 | — |
Experimental
The purity determinations of the products were accomplished by TLC on silica gel polygram STL G/UV 254 plates. The melting points of products were determined with an Electrothermal Type 9100 melting point apparatus. The FT-IR spectra were recorded on an Avatar 370 FT-IR Therma Nicolet spectrometer. The NMR spectra were provided on Brucker Avance 100 and 400 MHz instruments in acetone-d6, DMSO-d6 and CD3CN. Elemental analyses were performed using a Thermo Finnegan Flash EA 1112 Series instrument. Mass spectra were recorded with Agilent Technologies (HP) 5973 Network Mass Selective Detector and Shimadzu GC-MS-QP5050 instruments at 70 eV. Thermogravimetric analysis (TGA) was performed on a Shimadzu Thermogravimetric Analyzer (TG-50) under air atmosphere. BET surface area and pore size distribution were measured on a Belsorp mini II system at −196 °C using N2 as adsorbate. Transmission electron microscopy (TEM) examination was performed with a TEM microscope Leo 912 AB120 kV Zeiss Germany. Inductively coupled plasma (ICP) was carried out on a Varian, VISTA-PRO, CCD, Australia. Elemental compositions were determined with a Leo 1450 VP scanning electron microscope equipped with an SC7620 energy dispersive spectrometer (SEM-EDS) presenting a 133 eV resolution at 20 kV. All of the products were known compounds and characterized by the FT-IR spectroscopy and comparison of their melting points with known compounds. The structure of selected products was further confirmed by 1HNMR, 13CNMR spectroscopy, and mass spectrometry.Preparation of activated silica (II)
Silica (5.00 g, Merck grade 60) was heated at reflux with hydrochloric acid solution (37%, 30 mL). After 8 h the white suspension was filtered and the precipitate was thoroughly washed with deionized water until the pH of the eluent reached 6. The dried activated silica (I) was then treated with refluxing aqueous solution of NaOH (0.04 M, 30 mL) for 8 h. Then the suspension was filtered and washed with deionized water until a constant pH = 8 to afford a white solid powder (activated silica II) that was dried at 110 °C in an oven overnight under vacuum.Preparation of epoxy activated silica (III)
Activated silica (II) (2 g) was suspended to pure epichlorohydrin (10 mL) at 60 °C with vigorous stirring. After 24 h the resulting suspension was cooled to room temperature and centrifuged. The white precipitate was washed with methanol (4 × 15) until removing additional amount of epichlorohydrin. The epoxy activated silica (III) was then dried at 100 °C under vacuum for 8 h.Preparation of aminated epichlorohydrin activated silica (IV)
The epoxy activated silica (III) (2 g) was suspended in ammonia solution (25%, 20 mL) at 50 °C with stirring. After 72 h, the resulting suspension was cooled to room temperature and centrifuged. The resulting white powder was washed in turn with methanol (3 × 5) and water (4 × 15) until neutralizing the filtrate. The aminated epichlorohydrin activated silica (IV) was dried under vacuum at 100° C for 10 h.Preparation of Cu(II) immobilized on aminated epichlorohydrin silica (CAES)
To a solution of Cu(OAc)2·H2O (0.6 mmol, 0.11 g) in absolute EtOH (5 mL), aminated epichlorohydrin activated silica (IV) (1 g) was added at room temperature. The reaction mixture was stirred under Ar atmosphere for 4 h. The resulting suspension was centrifuged and the blue precipitate was washed in turn with acetone (4 × 5), ethanol (4 × 5) and then dried under vacuum at ambient temperature for 16 h.Preparation of CAES
As can be seen in Scheme 1, the catalyst was prepared in three steps. Initially, the surface of silica was activated by refluxing HCl (37%). Hydrochloric acid increased the terminal Si–OH (I, Scheme 1).44 The resulting precipitate was then treated with boiling NaOH solution which converted the Si–OH to negatively charged Si–O− groups to produce the desired activated silica (II, Scheme 1).44 The epoxy activated silica (III, Scheme 1) was obtained upon treatment of II with epichlorohydrin at 60 °C which under treatment with ammonia produced aminated epichlorohydrin activated silica (IV). The aminated epichlorohydrin activated silica was used as the support and ligand for entrapment of Cu(II) by the reaction of IV with a solution of Cu(OAc)2·H2O. The resulting complex contains Cu(II) immobilized on aminated epichlorohydrin activated silica matrix (CAES).Characterization of CAES
The catalyst was characterized by a battery of techniques including techniques: FT-IR spectroscopy, thermogravimetric analysis (TGA), transmission electron microscopy (TEM), scanning electron microscopy (SEM-EDS), BET analysis, Elemental analysis (CHN) and inductively coupled plasma (ICP).The catalyst structure was defined by FT-IR spectroscopy. Infrared data are useful only to confirm the existence of the bonded species. Fig. 1 illustrates the FT-IR spectra of activated silica with HCl(I), activated silica with NaOH(II), epoxy activated silica (III), aminated epichlorohydrin activated silica (IV) and Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). The principal bands for activated silica with HCl were observed at 1064, 808 and 469 cm−1 which are presumably due to asymmetric, symmetric and bending vibrations of Si–O–Si respectively. Also, strong and broad absorption band around 3475 cm−1 confirmed the presence of the hydrogen bonded Si–OH groups and adsorbed water which was disappeared in the FT-IR spectrum of activated silica with NaOH(II).
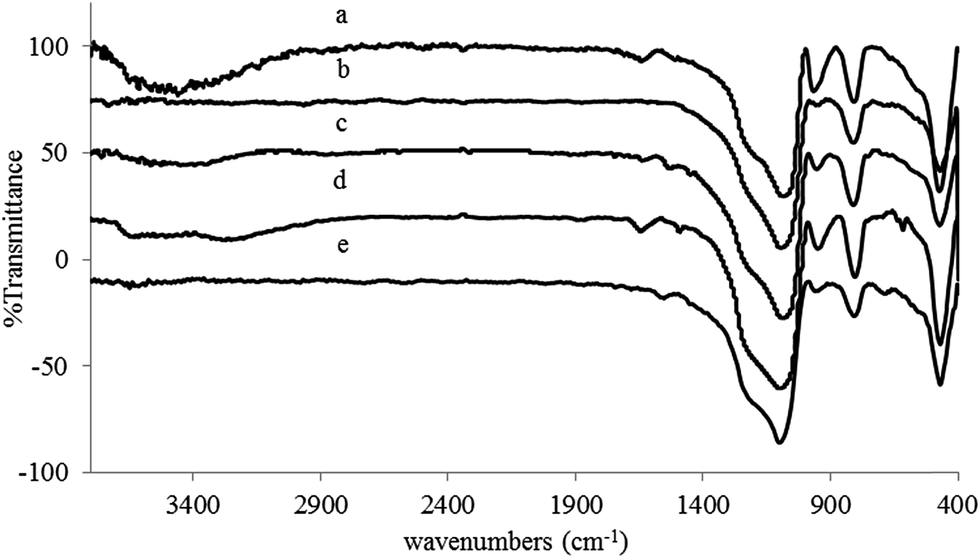 | ||
| Fig. 1 FT-IR spectrum of (a) activated silica with HCl(I); (b) activated silica with NaOH solution (II); (c) epoxy activated silica (III); (d) aminated epichlorohydrin activated silica (IV); (e) Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
Due to the low concentration of the organic part of modifier on the surface of silica (see FT-IR spectrum of III), the intensity of the new bands attesting the presence of organic groups is weak.45 The epoxy rings which are attached to the silicon framework are identified by the methylene C–H stretching bands46 at roughly 3050 cm−1 and C–O–C vibrational stretching at 1260–1240 cm−1. The latter vibration frequency was covered by the broad characteristic band of asymmetric vibration of Si–O–Si. Consequently, the asymmetric vibration band of Si–O–Si was appeared as a more broadened absorption band. The results showed that the silica surface has been immobilized by covalent bonded organic epoxy rings. Ring opening of epoxy ring with ammonia was confirmed by appearance of a broad absorption band around 3670–2900 cm−1 attributed to –OH and –NH2 stretching frequencies and furthermore, other absorption bands at 1644, 1245 and 1100 cm−1 due to –NH bending vibration, C–N and C–O stretching vibrations respectively. The two latter absorption bands were covered by asymmetric vibration band of Si–O–Si.
Fig. 2 shows the expanded FT-IR spectrum of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). Absorption bands at 428, and 450 cm−1 corroborate the coordination of Cu(II) on aminated epichlorohydrin activated silica which is attributed to Cu–N and Cu–O vibrations respectively. The latter absorption band was covered by bending vibration frequency of Si–O–Si. Upon coordination of Cu(II), the intensities of –NH2 stretching frequencies (3624 and 3251 cm−1) were decreased drastically. The –NH bending absorption band was also shifted to lower frequency upon coordination with Cu(II).
 | ||
| Fig. 2 FT-IR spectrum (expanded) of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
The TGA of aminated epichlorohydrin activated silica (IV) (Fig. 3) shows two main weight losses. The first one (2%, 27–300 °C) is accounted for physically and hydrogen bonded water in the structure of IV. The second one which is occurred at 300–560 °C (11%) is related to the decomposition of organic segment anchored to the silica surface. This part of the thermogram reveals the amounts of aminated epichlorohydrin on silica which is estimated to be 11% (w/w). So, the elevated temperature for pendent organic group removal, indicates the high thermal stability of IV. Thus, the high thermal stability of IV confirms the covalent bonding of the aminated epichlorohydrin groups on the surface of silica.
 | ||
| Fig. 3 TGA Thermogram of aminated epichlorohydrin activated silica (IV). | ||
In addition to structural confirmation and thermogravimetric analysis, quantitative determination of covalently bonded epoxy ring and aminated epoxy ring onto the surface of silica (III, IV) was performed by elemental analysis. The elemental analysis of III showed the carbon content to be 4.316%. Also, the results of elemental analysis of IV which are in good agreement with those of the results of thermogravimetric analysis, showed that carbon and nitrogen contents of IV were found to be 4.316% and 0.35% respectively which revealed that 1.15 mmol of aminated epichlorohydrin was incorporated into the 1.000 g of IV.
The copper content of CAES was obtained by inductively coupled plasma (ICP) to be 1.6092 wt%, 0.0158 g, 0.25 mmol of Cu per 1.000 g of CAES.
The TEM micrographs and size distributions (40 and 32 nm) of nanocatalyst Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES) is shown in Fig. 4. TEM images indicated that the most of the prepared nanoparticles are spherical shaped and have sized 10–20 nm.
 | ||
| Fig. 4 TEM micrographs of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
Scanning electron microscopy (SEM) revealed that the nanocatalyst presented the uniform particles with spherical morphology (Fig. 5) which are in good agreement with the results of TEM. Also, it could be seen that the nanocatalyst has not been destroyed during the modifications. The energy dispersive spectrum (EDS) indicated the presence of C, O, Si and Cu elements (Fig. 6). This analysis confirms that Cu(II) is supported on aminated epichlorohydrin activated silica (IV).
 | ||
| Fig. 5 SEM micrographs of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
 | ||
| Fig. 6 The EDS spectrum of Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). | ||
Table 5 gives the specific surface area and the mean pore diameter data of the activated silica with HCl(I), aminated epichlorohydrin activated silica (IV) and Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). From the nitrogen adsorption–desorption isotherms and BET analysis (see ESI†) the specific surface area of I, IV and CAES were calculated as 362, 346 and 329 m2 g−1 respectively. Then, we can conclude that all modified samples exhibited similar isotherm shape which suggested no obvious change of structure after modification. As can be seen from the data obtained from Table 5, functionalization of activated silica by aminated epichlorohydrin and immobilization of Cu(II) on aminated epichlorohydrin activated silica decreased the specific surface area.
| Sample | SBET (m2 g−1) | Mean pore diameter (nm) | ICP (wt%) |
|---|---|---|---|
| I | 362 | 7.139 | — |
| IV | 346 | 8.403 | — |
| CAES | 329 | 8.57 | 1.6092 |
Typical procedure for the preparation of 5-phenyl-1H-tetrazole
Sodium azide (0.0650 g, 1 mmol) and CAES (0.0400 g, 1 mol%), were added to a solution of benzonitrile (0.1031 g, 1 mmol) in DMSO (2 mL) with stirring at room temperature. The reaction temperature was raised up to 130 °C for 1 h. The progress of the reaction was monitored by TLC. After completion of reaction, the reaction mixture was cooled and centrifuged. The filtrate was treated with HCl (4 N, 10 mL) and then EtOAc (10 mL). The resultant organic layer was separated, washed with distilled water (2 × 10), dried over anhydrous sodium sulfate, and concentrated to give the crude solid 5-phenyl-1H-tetrazole. The crude product was recrystallized from n-hexane/ethylacetate (1:1) obtaining 0.1386 g of colourless crystals (95% yield).
Conclusion
In conclusion, a simple and effective method for the synthesis of 5-substituted-1H-tetrazoles from nitriles and sodium azide has been developed using nanocatalyst Cu(II) immobilized on aminated epichlorohydrin activated silica (CAES). This nano sized heterogeneous catalyst is easily prepared and stable to air which shows excellent catalytic performance for various nitriles. Also, the nanocatalyst could be separated easily from the reaction mixture which offers simple work-up and high yields. The catalytic activity of the catalyst remains unaltered after five consecutive cycles. The given methodology is efficient and environmentally benign.Acknowledgements
The authors gratefully acknowledge the partial support of this study by Ferdowsi University of Mashhad Research Council (Grant no. p/3/32111).Notes and references
- (a) R. N. Butler, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, UK, 1996, vol. 4, p. 674 Search PubMed; (b) H. Singh, A. S. Chala, V. K. Kapoor, D. Paul and R. K. Malhotra, Prog. Med. Chem., 1980, 17, 151 CrossRef CAS; (c) V. A. Ostrovskii, M. S. Pevzner, T. P. Kofmna, M. B. Sheherbinin and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 CAS; (d) M. Hiskey, D. E. Chavez, D. L. Naud, S. F. Son, H. L. Berghout and C. A. Bome, Proc. Int. Pyrotech. Semin., 2000, 27, 3 Search PubMed.
- D. M. Zimmerman and R. A. Olofson, Tetrahedron Lett., 1969, 10, 5081 CrossRef.
- M. Rahman, A. Roy, M. Ghosh, Sh. Mitra, A. Majee and A. Hajra, RSC Adv., 2014, 4, 6116 RSC.
- D. R. Patil, Y. B. Wagh, P. G. Ingole, K. Singh and D. S. Dalal, New J. Chem., 2013, 37, 3261 RSC.
- L. Bosch and J. Vilarrasa, Angew. Chem., 2007, 46, 3926 CrossRef CAS PubMed.
- G. Venkateshwarlu, A. Premalatha, K. C. Rajanna and P. K. Saiprakash, Synth. Commun., 2009, 39, 4479 CrossRef CAS.
- J. Bonnamour and C. Bolm, Chem.–Eur. J., 2009, 15, 4543 CrossRef CAS PubMed.
- D. J. Carini, J. V. Duncia, P. E. Aldrich, A. T. Chiu, A. L. Johnson, M. E. Pierce, W. A. Price, J. B. Santella and G. J. Wells, J. Med. Chem., 1991, 34, 2525 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS PubMed.
- (a) Z. P. Demko and K. B. Sharpless, Org. Lett., 2002, 4, 2525 CrossRef CAS PubMed; (b) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210 CrossRef CAS PubMed; (c) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983 CrossRef CAS PubMed.
- P. Mani, A. K. Singh and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 1879 CrossRef CAS PubMed.
- (a) J. Roh, T. V. Atramonova, K. Vavrova, G. I. Koldobskii and A. Hrabalek, Synthesis, 2009, 2175 CrossRef CAS PubMed; (b) K. Koguro, T. Oga, S. Mitsui and R. Orita, Synthesis, 1998, 910 CrossRef CAS PubMed.
- S. K. Prajapti, A. Nagarsenkar and B. N. Babu, Tetrahedron Lett., 2014, 55, 3507 CrossRef CAS PubMed.
- D. P. Matthews, J. E. Green and A. J. Shuker, J. Comb. Chem., 2000, 2, 19 CrossRef CAS PubMed.
- A. Kumar, R. Narayanan and H. Shechter, J. Org. Chem., 1996, 61, 4462 CrossRef CAS.
- M. Nasrollahzadeh, Y. Bayat, D. Habibi and S. Moshaee, Tetrahedron Lett., 2009, 50, 4435 CrossRef CAS PubMed.
- D. Amantini, R. Beleggia, F. Fringuelli, F. Pizzo and L. Vaccaro, J. Org. Chem., 2004, 69, 2896 CrossRef CAS PubMed.
- V. S. Patil, K. P. Nandre, A. U. Borse and S. V. Bhosale, E-J. Chem., 2012, 9, 1145 CrossRef CAS PubMed.
- B. Das, C. R. Reddy, D. N. Kumar, M. Krishnaiah and R. Narender, Synlett, 2010, 391 CrossRef CAS PubMed.
- S. J. Wittenberger and B. G. Donner, J. Org. Chem., 1993, 58, 4139 CrossRef CAS.
- (a) S. H. Watterson, Z. Xiao, D. S. Dodd, D. R. Tortolani, W. Vaccaro, D. Potin, M. Launay, D. K. Stetsko, S. Skala and P. M. Davis, J. Med. Chem., 2010, 53, 3814 CrossRef CAS PubMed; (b) S. Takale, S. Manave, K. Phatangare, V. Padalkar, N. Darvatkar and A. Chaskar, Synth. Commun., 2012, 42, 2375 CrossRef CAS; (c) Z. Du, C. Si, Y. Li, Y. Wang and J. Lu, Int. J. Mol. Sci., 2012, 13, 4696 CrossRef CAS PubMed.
- A. Khalafi-Nezhad and S. Mohammadi, RSC Adv., 2013, 3, 4362 RSC.
- J. He, B. Li, F. Chen, Z. Xu and G. Yin, J. Mol. Catal. A: Chem., 2009, 304, 135 CrossRef CAS PubMed.
- J. Braun and W. Keller, Ber. Dtsch. Chem. Ges., 1932, 65, 1677 CrossRef.
- (a) L. Lang, B. Li, W. Liu, L. Jiang, Z. Xu and G. Yin, Chem. Commun., 2010, 46, 448 RSC; (b) L. Lang, H. Zhou, M. Xue, X. Wang and Z. Xu, Mater. Lett., 2013, 106, 443 CrossRef CAS PubMed.
- M. L. Kantam, K. B. Shiva Kumar and K. Phani Raja, J. Mol. Catal. A: Chem., 2006, 247, 186 CrossRef CAS PubMed.
- M. Lakshmi Kantam, K. B. S. Kumar and C. Sridhar, Adv. Synth. Catal., 2005, 347, 1212 CrossRef.
- S. M. Agawane and J. M. Nagarkar, Catal. Sci. Technol., 2012, 2, 1324 CAS.
- G. Aridoss and K. K. Laali, Eur. J. Org. Chem., 2011, 2011, 6343 CrossRef CAS.
- M. L. Kantam, V. Balasubrahmanyam and K. B. Shiva Kumar, Synth. Commun., 2006, 36, 1809 CrossRef CAS.
- Y. S. Gyoung, J.-G. Shim and Y. Yamamoto, Tetrahedron Lett., 2000, 41, 4193 CrossRef CAS.
- T. Jin, F. Kitahara, S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 2824 CrossRef CAS PubMed.
- P. Mani, C. Sharma, S. Kumar and S. K. Awasthi, J. Mol. Catal. A: Chem., 2014, 392, 150 CrossRef CAS PubMed.
- D. Kong, Y. Liu, J. Zhang, H. Li, X. Wang, G. Liu, B. Li and Z. Xu, New J. Chem., 2014, 38, 3078 RSC.
- F. Dehghani, A. R. Sardarian and M. Esmaeilpour, J. Organomet. Chem., 2013, 743, 87 CrossRef CAS PubMed.
- B. Sreedhar, A. S. Kumar and D. Yadav, Tetrahedron Lett., 2011, 52, 3565 CrossRef CAS PubMed.
- (a) D. P. Curran, S. Hadida and S. Y. Kim, Tetrahedron, 1999, 55, 8997 CrossRef CAS; (b) R. Shelkar, A. Singh and J. Nagarkar, Tetrahedron Lett., 2013, 54, 106 CrossRef CAS PubMed.
- B. Franca, C. Luca, M. Raimondo and S. Giovanni, J. Org. Chem., 1999, 64, 1033 CrossRef.
- S. Shanmuganathana, L. Greiner and P. D. de Maríaa, Tetrahedron Lett., 2010, 51, 6670 CrossRef PubMed.
- (a) M. H. Valkenberg and W. F. Holderich, Catal. Rev.: Sci. Eng., 2002, 44, 321 CrossRef CAS PubMed; (b) E. L. Margelefsky, R. K. Zeidan and M. E. Davis, Chem. Soc. Rev., 2008, 37, 1118 RSC; (c) J. C. Hicks, R. Dabestani, A. C. Buchanan and C. W. Jones, Chem. Mater., 2006, 18, 5022 CrossRef CAS; (d) J. C. Hicks, R. Dabestani, A. C. Buchanan and C. W. Jones, Inorg. Chim. Acta, 2008, 361, 3024 CrossRef CAS PubMed; (e) M. W. McKittrick and C. W. Jones, J. Am. Chem. Soc., 2004, 126, 3052 CrossRef CAS PubMed; (f) K. Q. Yu, M. W. McKittrick and C. W. Jones, Organometallics, 2004, 23, 4089 CrossRef CAS.
- J. F. Fritz and J. N. King, Anal. Chem., 1976, 48, 570 CrossRef CAS.
- B. Akhlaghinia and S. Tavakoli, Synthesis, 2005, 1775 CrossRef CAS PubMed.
- B. Akhlaghinia and S. Rezazadeh, J. Braz. Chem. Soc., 2012, 23, 2197 CrossRef CAS PubMed.
- H. Sharghi, M. H. Beyzavi and M. M. Doroodmand, Eur. J. Org. Chem., 2008, 24, 4126 CrossRef.
- Special Issue, V. Coman, R. Grecu, J. Wegmann, S. Bachmann and K. Albert, Stud. Univ. Babes-Bolyai, Phys., 2001, 326–331 Search PubMed.
- F. Adam, K. M. Hello and H. Osman, Chin. J. Chem., 2010, 28, 2383 CrossRef CAS.
- V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 46, 8440 CrossRef CAS PubMed.
- S. Lossen and J. Liebigs, Ann. Chem., 1897, 298, 102 Search PubMed.
- V. Rama, K. Kanagaraj and K. Pitchumani, J. Org. Chem., 2011, 76, 9090 CrossRef CAS PubMed.
- A. Köennecke, R. Döme, E. Kleinpeter and E. Lippmann, Tetrahedron, 1997, 35, 1957 CrossRef.
- F. Ek, L. G. Wistrand and T. Frejd, J. Org. Chem., 2003, 68, 1911 CrossRef CAS PubMed.
- Z. Yizhong, R. Yiming and C. Chun, Helv. Chim. Acta, 2009, 92, 171 CrossRef.
- G. Satzinger and J. Liebigs, Ann. Chem., 1960, 638, 159 CAS.
- A. Najafi Chermahini, A. Teimouri and A. Moaddeli, Heteroat. Chem., 2011, 22, 168 CrossRef.
- A. R. Katritzky, B. E. M. El-Gendy, B. Hall, C. D. Draghici and P. J. Steel, J. Org. Chem., 2010, 75, 6468 CrossRef CAS PubMed.
- M. Herbst, J. Org. Chem., 1950, 15, 1082 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra15148h |
| This journal is © The Royal Society of Chemistry 2015 |