
Length-dependent rectification and negative differential resistance in heterometallic n-alkanedithiol junctions
Jian Shaoad,
X. Y. Zhangacd,
Yue Zheng*abd,
Biao Wang*ac and
Yun Chenad
aState Key Laboratory of Optoelectronic Materials and Technologies, School of Physics and Engineering, Sun Yat-sen University, 510275, Guangzhou, China. E-mail: zhengy35@mail.sysu.edu.cn; Tel: +86-20-8411-3231
bDepartments of Mechanical Engineering and Civil and Environmental Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: yue.zheng@northwestern.edu; Tel: +1-224-999-5961
cSino-French Institute of Nuclear Engineering and Technology, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: wangbiao@mail.sysu.edu.cn; Tel: +86-20-8411-5692
dMicro&Nano Physics and Mechanics Research Laboratory, School of Physics and Engineering, Sun Yat-sen University, 510275, Guangzhou, China. E-mail: zhengy35@mail.sysu.edu.cn; Tel: +86-20-8411-3231
First published on 14th January 2015
Abstract
The transport properties of heterometallic n-alkanedithiol junctions have been investigated via first-principles calculations. Results show that the heterometallic n-alkanedithiol junctions exhibit significant rectification at lower voltage. A negative differential resistance was found at higher voltage, which increases with the increase of the n-alkanedithiol backbone length. In order to explain these phenomena, the molecular orbitals of n-alkanedithiol have been analyzed between certain electrodes. It is found that the rectification is induced by asymmetric orbital profiles between the heterometallic electrodes, and negative differential resistance arises when the molecular orbitals cross the band edge provided by the metal–sulfur bond.
Introduction
Molecular electronic devices1 are attracting booming interest for their inherent advantages in both size and bio-affinity over their inorganic counterparts.2 Single-molecule junctions3,4 are the most basic component of molecular devices. Interesting transport phenomena have been observed in single-molecule junctions, such as rectification5,6 and negative differential resistance7 (NDR). These effects are fundamental and promising for building single molecular circuits. Rectification in single-molecule junctions can be induced by two mechanisms, i.e. the donor–σ–acceptor system and the asymmetric couplings of the molecule to the electrodes.5,8,9 Previous studies have been more focused on rectification induced by the asymmetry of a central molecule.10,11 Meanwhile, investigations into the effect of asymmetric electrodes on the molecular rectification have just begun. Typical works, such as that by Chen et al.,12 have exhibited an obvious rectification in symmetric molecule bridged Au/Pt nanowire heterojunctions. Dalgleish et al.13 investigated 4-alkanedithiol junctions with heterometallic electrodes via a semi-empirical extended-Huckel model,14 and found rectification in the junctions due to a lack of d-orbitals near the Fermi level in one of the electrodes. Moreover, some investigations15 have also suggested that molecular junctions with the rectification effect often show combined NDR, which is invoked by resonant tunneling when the molecule is weakly coupled to the electrode.16It is well known that the transport properties of single molecular junctions17–20 are determined by the level alignment and localization of molecular orbitals.21 Meanwhile, electrodes play an important role by coupling with the molecular orbital. Compared with symmetric electrodes, heterometallic electrodes22 induce asymmetric hybridizations in opposite interfaces and can bring additional controllability to molecular junctions. Due to the complexity of the molecule–electrode interaction, the interface should be scrutinized in detail. Moreover, the different work functions of the heterometallic electrodes produce a built-in electric field.23 For a molecule of ∼nanometers, this electric field is large enough to shift the molecular levels and may result in new characteristics of the junction. So far, it is still unknown how the molecular orbital reacts to the built-in electric field and how much this effect can alter the current.24 In addition, because the distribution of electrons in the molecule is highly restricted and anisotropic, the localization of orbitals should be strongly dependent on the length of the bridging molecule. Until now, however, how the length of the molecule will affect transport properties like rectification and NDR in single-molecule junctions has not been fully investigated. Thus a comprehensive study is required to clarify the problems.
In this paper, the bias dependent transport properties of heterometallic n-alkanedithiol junctions with different backbone lengths have been calculated via first-principles simulations. A significant rectification of heterometallic n-alkanedithiol junctions has been found. The NDR effect has also been observed, and is dependent on the backbone length of the molecule. To explain the rectification and NDR phenomena, the influence of electrodes on the molecular orbitals of the junctions has been discussed, which puts an emphasis on the coupling in the molecule–electrode interface and the built-in electric field through the scattering region.
Method
In our simulation, the alkanedithiol molecule was first optimized in the gas phase and then connected to the adatoms on seven layers of metal atoms, as shown in Fig. 1. All combinations of commensurate Au/Ag/Al asymmetric heterometallic electrodes for the junctions have been investigated, respectively. The density functional26,27 calculations were performed within the generalized gradient approximations of Perdew–Burke–Ernzerhof (GGA-PBE).28 Structural relaxations were implemented in the VASP code.29 A plane wave basis set with an energy cutoff of 450 eV based on a projector augmented wave (PAW) method was utilized. Γ-point Brillouin zone integration was used for relaxation until the force on each atom reached the tolerance limit of 0.05 eV Å−1. | ||
| Fig. 1 Schematic illustration of the atomic structure of Au/n-alkanedithiol/Ag heterometallic junctions (n = 2, 4, 6, 8, 10, 12). Atomic species are shown in yellow for Au, silver for Ag, red for S, black for C, and white for H. The whole scattering region is shown and the two glassy boxes indicate the unit cells of the periodic half-infinite structure. | ||
After relaxation, transport calculations were performed within a density functional theory (DFT) based non-equilibrium Green function (NEGF) approach25 using an Atomistix ToolKit.30,31 Here an energy cutoff of 75 Rydberg was used in the framework of a linear combination of atomic orbitals (LCAO) basis set. All atoms in the molecule were modeled with double-ζ plus polarization basis sets and the metal atoms were modeled with single-ζ plus polarization basis sets for efficiency. Self-consistency was achieved using 4 × 4 k-point sampling in the two-dimensional Brillouin zone. Then the energy resolved transmission spectra were calculated respectively at different voltages with an 8 × 8 k-mesh. Local density of states (LDOS) with a 21 × 21 k-mesh was averaged in the x–y plane to investigate its variation along the z axis. In addition, all the energies in the following sections were calculated relative to the Fermi level.
Results and discussion
The transport properties of Au/n-alkanedithiol/Ag junctions
To illustrate the transport properties of Au/n-alkanedithiol/Ag junctions (‘Cn’, n = 2, 4, 6, 8, 10, 12, marking the number of carbon atoms in backbone), the I–V curves have been plotted in Fig. 2(a). For n ≥ 4, the molecular length dependence of the current is verified by fitting to the general expression I ∝ exp(−βn). The concatenate fit value of β is 0.93 ± 0.15 per methylene (0.75 ± 0.12 Å−1), similar to previous experimental results (0.80 ± 0.08 Å−1).32 Inside a ±1.0 V range, the absolute current is asymmetric. The I–V relationship is in accordance with a Simmons model23 with a voltage-dependent barrier height. At a larger positive voltage, significant NDR can be observed, i.e., the current decreases as the voltage increases in the range of +1.1 V to +2.0 V. Inside the range of −1.1 V to −2.0 V, however, the currents appear to be stable. In addition, the conductance of the Au/8-alkanedithiol/Ag junction at zero bias (35 nS) is located between the Au/8-alkanedithiol/Au junction (20 nS) and the Ag/8-alkanedithiol/Ag junction (50 nS). For the C2 junction, the current is obviously larger compared with a Simmons model over the range of −0.2 V to −0.6 V, and thus shows NDR in the −0.6 V to −1.0 V range. This phenomenon is caused by a different transport mechanism, which will be discussed later. For voltages outside a ±1.0 V range, the electric field applied on a C2 junction was too strong and destabilized the molecule, so we discarded these data. In summary, all Au/n-alkanedithiol/Ag junctions show asymmetric I–V characteristics under an electric field with opposite directions. For comparison, no asymmetric current or NDR has been found in symmetric Au/n-alkanedithiol/Au junction up to ±3 V according to Cui et al.33 | ||
| Fig. 2 (a) The absolute currents of Cn junctions with n = 2, 4, 6, 8, 10, 12 versus voltages (upper panel) and a fitted slope, β, in I ∝ exp(−βn) for Cn junctions with n = 4–12, together with error bars at each voltage (lower panel). (b) The I+/I− versus absolute voltage of Cn junctions with n = 2–12. | ||
The asymmetry of the I–V characteristics has been further illustrated by the rectification ratio I+/I− with voltage dependence (see Fig. 2(b)). It is found that the rectification ratio is dependent on both the voltage and molecular length. The rectification ratio of the Cn junction with n ≥ 4 increases to as much as 2 at 1 V with an identical slope, and it decreases in the range of 1 V to 2 V with the effect of NDR. It is also found that the NDR is clearly dependent on molecular length because the rectification ratios reduce more steeply with the increase of molecular length. In contrast, the rectification ratio in the C2 junction is much smaller than the others, indicating a critical length effect. Specifically, the rectification ratio oscillates until the voltage reaches 0.7 V and then increases obviously in the 0.7 V to 1.0 V range. The valley near 0.5 V is induced by an obscure current increase in the range of −0.2 V to −0.7 V. Similar results have also been observed in junctions with Au/Al and Ag/Al electrodes.
The local density of states and molecular projected self-consistent Hamiltonian levels
In order to explain the rectification in Cn junctions with n ≥ 4, the molecular projected self-consistent Hamiltonian (MPSH) levels of the heterometallic junctions have been analyzed. It is found that the degenerate molecular energy levels split up to 0.5 eV due to heteroelectrodes. This energy level split is introduced by two asymmetry factors: first, the asymmetric hybridization of degenerate molecular orbitals with the left and right electrodes; second, the built-in electric field. The first factor of the C8 junction has been visualized in the LDOS along the transport direction, as shown in Fig. 3(a). It is found that the extra 5d orbital in Au compared with Ag leads to asymmetric local density of surface states in the −2.5 eV to −1.5 eV range, i.e., high density for an Au–S bond (part 3 in Fig. 3(a)) and low density for an Ag–S bond (part 4 in Fig. 3(a)). Thus the degenerate orbitals hybridize their energy levels to the asymmetric surface states and split into MPSH eigenstates A and B (shown in red and blue dashed lines in Fig. 4(b)). Meanwhile, the MPSH eigenstates A and B are broadened into the density of states (DOS), Di(E) = (γi/2π)/[(E − Ei)2 + (γi/2)2], where γi(E) = γiL(E) + γiR(E), γiL and γiR are the self-energy (or the coupling) of level i = A or B due to left and right electrodes, respectively (Table 1). As a consequence, the C2h symmetry of the molecule is broken due to the connection with the heterometallic electrodes. On the other hand, the built-in electric field is induced by the difference in the work functions of the two electrodes. In Au/Ag heteroelectrodes, the difference is ∼1 eV. As illustrated in Fig. 4(a), the MPSH eigenstates A and B are separated by the insulating saturate bonds and are localized respectively onto the left and right ends of the molecule. So the built-in electric field produces a voltage drop and realigns the energy levels of the two states (shown in red and blue solid lines in Fig. 4(b)). Due to these two factors, the junctions will exhibit rectification under bias. This is because the MPSH levels of A and B will further shift ±0.45eVb in opposite directions under applied voltage Vb as illustrated in Fig. 4(c). The energy resolved transmission spectra are plotted in Fig. 4(d). It is shown that the peaks are in correspondence with the MPSH eigenstates. According to the Landauer–Büttiker (LB) formula,34–37 only the transmission within the bias window over −eVb/2 to eVb/2, dashed lines in Fig. 4(d), contributes to the current . It is found that the peak induced by MPSH eigenstate B locates inside the bias window at positive voltages while no peak goes into the bias window at negative voltages. As a result, the current under a positive voltage is larger than that under a negative voltage and the junction rectifies. The rectification can also be fitted to a Simmons model23 if the transmission barrier height is defined as ΦB(Vb) = ΦB(0) − 0.45eVb. For other asymmetric electrodes like Au/Al, asymmetric movements of energy levels can also be observed (see Table 2) and rectification can be expounded in similar ways.
. It is found that the peak induced by MPSH eigenstate B locates inside the bias window at positive voltages while no peak goes into the bias window at negative voltages. As a result, the current under a positive voltage is larger than that under a negative voltage and the junction rectifies. The rectification can also be fitted to a Simmons model23 if the transmission barrier height is defined as ΦB(Vb) = ΦB(0) − 0.45eVb. For other asymmetric electrodes like Au/Al, asymmetric movements of energy levels can also be observed (see Table 2) and rectification can be expounded in similar ways.
| Eigenstate | Composition | +1 V (eV) | −1 V (eV) |
|---|---|---|---|
| Eigenstate A | Au–S–(CH2)8 | −2.00 | −1.18 |
| Eigenstate B | (CH2)8–S–Ag | −0.60 | −1.52 |
| Eigenstate C | Au–S–CH2 | −1.30 | −0.38 |
| Eigenstate D | CH2–S–Ag | −0.09 | −0.85 |
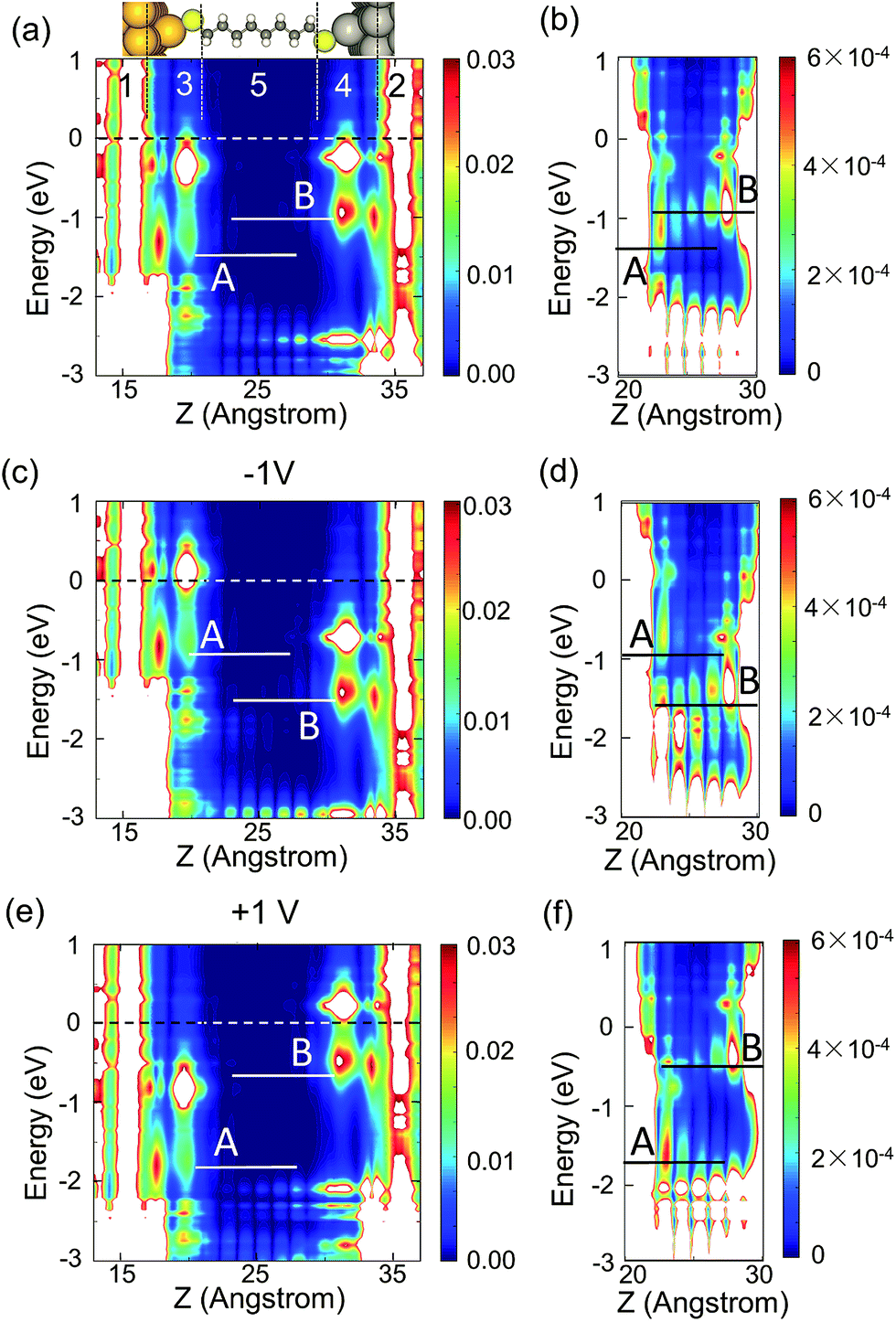 | ||
| Fig. 3 (a) LDOS at equilibrium averaged in the x–y plane and the atomic structure of the C8 junction in real space with the same z axis. The white area shows LDOS beyond the upper limit of the color bar (very high DOS). The dashed line indicates the average Fermi level. LDOS in part 5 is plotted aside in (b) with a fiftieth contour value of (a). For −1 V and +1 V, the LDOS in the scattering region and part 5 are shown in (c)–(f) respectively. | ||
 | ||
| Fig. 4 (a) MPSH eigenstates (0.02 absolute contour cleave) of the C8 junction marked as A, B, C and D. (b) Schematic illustration of molecular level movement during bridging between heterometallic electrodes at equilibrium. The black solid line indicates the potential energy drop caused by the built-in electric field. (c) The energy level movements of MPSH eigenstates A (red) and B (blue) from −1 V to +1 V. (d) The energy resolved transmission spectra of the C8 junction under bias. | ||
| Eigenstate | Composition | +1 V (eV) | −1 V (eV) |
|---|---|---|---|
| Eigenstate E | Au–S–(CH2)8 | −2.17 | −1.28 |
| Eigenstate F | (CH2)8–S–Al | −1.99 | −2.77 |
| Eigenstate G | Au–S–CH2 | −1.33 | −0.41 |
| Eigenstate H | CH2–S–Al | −1.03 | −1.84 |
Our second important discovery is the length dependent NDR effect found in the +1.1 V to +2 V range. The arising of NDR at +1 V for all heterojunctions in this work is caused by the crossing of MPSH level B through the band edge provided by an Au–S bond (see Fig. 3(e) and (f)). Interestingly, the peak–valley ratio of NDR increases with the increase of the n-alkanedithiol backbone length (see Fig. 2). This is because the coupling of the molecular orbital with the electrode is strongly dependent on its localization. As the molecule becomes longer, the MPSH eigenstate B remains localized on the right end of the molecule and thus its coupling with the left electrode is weaker. According to the expression of Di(E), a smaller coupling leads to a sharper DOS broadening and consequently a steeper NDR effect. On the contrary, no significant NDR is observed at −1 V because MPSH level A locates inside the band provided by the Ag–S bond (see Fig. 3(c) and (d)).
Length-dependent rectification in heterometallic n-alkanedithiol junctions
Another interesting length-dependent effect is the transition in the transport mechanism from C2 to longer junctions. For longer junctions, the transport is dominated by the tunneling through MPSH eigenstates A and B and the contributions from MPSH eigenstates C and D are ignorable. For C2 junction, however, contributions from MPSH eigenstates C′ and D′ can modify the current significantly. It is found in the C2 junction that MPSH eigenstates C′ and D′ become delocalized through the backbone (see Fig. 5(a)), compared with localized MPSH eigenstates C and D in longer junctions (for example C8 junction in Fig. 4(a)). As shown in Fig. 5(b), this delocalization leads to new peaks in the energy resolved transmission spectra. Since the peak invoked by MPSH eigenstate D′ locates near the Fermi level, it dominates the current under bias. So the C2 junction shows different I–V and rectification characteristics from longer junctions (see Fig. 2(a) and (b)). | ||
| Fig. 5 (a) MPSH eigenstates (0.02 absolute contour cleave) of the C2 junction marked as A′, B′, C′ and D′. (b) The energy resolved transmission spectra at equilibrium of the Cn junctions (n = 2, 4, 6, 8). | ||
Conclusions
In summary, we have presented that n-alkanedithiol bridged heterometallic junctions exhibit length-dependent rectification and NDR effects via DFT calculations. The rectification is explained by the molecular level split due to the asymmetric hybridizations with the electrodes and the built-in electric field caused by a difference in the electrodes’ work functions. The rectification ratio of 2 is by no means large, however one should be aware that the junction is bridged by symmetric n-alkanedithiols. Besides, unlike Au/Pt electrodes, the Au/Ag electrodes are in the same group and have similar bonding energies with thiol. Thus the asymmetric properties come mainly from explicit electronic structure at the interface. Significant improvement can be made by combination with appropriate asymmetric molecules or conformations in the interface. In addition, length-dependent NDR in single-molecule junctions has been observed. The mechanisms of NDR and length-dependent rules are also investigated to give essential insight and reference for regulating heterometallic n-alkanedithiol junctions. By considering the asymmetric nature, our results could be helpful to reveal the electrodes’ ability to manipulate single molecular devices.Acknowledgements
The authors gratefully acknowledge the financial support of NSFC (nos 11402312, 51172291, 11232015, 11474363). Yue Zheng also thanks the support from the Fundamental Research Funds for the Central Universities to Micro&Nano Physics and Mechanics Research Laboratory, NCET in University, the Research Fund for the Doctoral Program of Higher Education, and Fok Ying Tung Foundation, the Science and Technology Innovation Project of Guangdong Provincial Education Department, Guangdong Natural Science Funds for Distinguished Young Scholar and the China Scholarship Council.Notes and references
- A. Nitzan and M. A. Ratner, Science, 2003, 300, 1384 CrossRef CAS PubMed.
- H. Hakkinen, Nat. Chem., 2012, 4, 443 CrossRef PubMed.
- M. Elbing, R. Ochs, M. Koentopp, M. Fischer, C. von Hanisch, F. Weigend, F. Evers, H. B. Weber and M. Mayor, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8815 CrossRef CAS PubMed.
- S. Y. Quek, L. Venkataraman, H. J. Choi, S. G. Louie, M. S. Hybertsen and J. B. Neaton, Nano Lett., 2007, 7, 3477 CrossRef CAS PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277 CrossRef CAS.
- N. J. Geddes, J. R. Sambles, D. J. Jarvis, W. G. Parker and D. J. Sandman, Appl. Phys. Lett., 1990, 56, 1916 CrossRef CAS PubMed.
- J. Chen, M. A. Reed, A. M. Rawlett and J. M. Tour, Science, 1999, 286, 1550 CrossRef CAS.
- J. Taylor, M. Brandbyge and K. Stokbro, Phys. Rev. Lett., 2002, 89, 138301 CrossRef.
- Y. Xue and M. Ratner, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115407 CrossRef.
- P. E. Kornilovitch, A. M. Bratkovsky and R. Stanley Williams, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 165436 CrossRef.
- I. Diez-Perez, J. Hihath, Y. Lee, L. Yu, L. Adamska, M. A. Kozhushner, I.I. Oleynik and N. Tao, Nat. Chem., 2009, 1, 635 CrossRef CAS PubMed.
- X. Chen, S. Yeganeh, L. Qin, S. Li, C. Xue, A. B. Braunschweig, G. C. Schatz, M. A. Ratner and C. A. Mirkin, Nano Lett., 2009, 9, 3974 CrossRef CAS PubMed.
- H. Dalgleish and G. Kirczenow, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 245431 CrossRef.
- R. Hoffmann, J. Chem. Phys., 1963, 39, 1397 CrossRef CAS PubMed.
- L. Esaki, Phys. Rev., 1958, 109, 603 CrossRef CAS.
- Y. Xue, S. Datta, S. Hong, R. Reifenberger, J. I. Henderson and C. P. Kubiak, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, R7852 CrossRef CAS.
- J. Reichert, R. Ochs, D. Beckmann, H. B. Weber, M. Mayor and H. von Lohneysen, Phys. Rev. Lett., 2002, 88, 4 Search PubMed.
- F. Chen, J. Hihath, Z. F. Huang, X. L. Li and N. J. Tao, in Annual Review of Physical Chemistry, Annual Reviews, Palo Alto, 2007, vol. 58, p. 535 Search PubMed.
- S. M. Lindsay and M. A. Ratner, Adv. Mater, 2007, 19, 23 CrossRef CAS.
- S. J. van der Molen and P. Liljeroth, J. Phys.: Condens. Matter, 2010, 22, 30 Search PubMed.
- S. Datta, Quantum transport: atom to transistor, Cambridge University Press, 2005 Search PubMed.
- J. A. Malen, S. K. Yee, A. Majumdar and R. A. Segalman, Chem. Phys. Lett., 2010, 491, 109 CrossRef CAS PubMed.
- J. G. Simmons, J. Appl. Phys., 1963, 34, 1793 CrossRef PubMed.
- V. B. Engelkes, J. M. Beebe and C. D. Frisbie, J. Am. Chem. Soc., 2004, 126, 14287 CrossRef CAS PubMed.
- Y. Q. Xue, S. Datta and M. A. Ratner, Chem. Phys., 2002, 281, 151 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- M. Brandbyge, J.-L. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 165401 CrossRef.
- M. S. José, A. Emilio, D. G. Julian, G. Alberto, J. Javier, O. Pablo and S.-P. Daniel, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef.
- B. Xu and N. Tao, Science, 2003, 301, 1221 CrossRef CAS PubMed.
- X. D. Cui, X. Zarate, J. Tomfohr, O. F. Sankey, A. Primak, A. L. Moore, T. A. Moore, D. Gust, G. Harris and S. M. Lindsay, Nanotechnology, 2002, 13, 5 CrossRef CAS.
- R. Landauer, IBM J. Res. Dev., 1957, 1, 223 CrossRef.
- R. Landauer, Philos. Mag., 1970, 21, 863 CrossRef CAS.
- M. Büttiker, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 9375 CrossRef.
- M. Büttiker, Ann. N. Y. Acad. Sci, 1990, 581, 176 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |