
Antioxidant vs. prooxidant action of phenothiazine in a biological environment in the presence of hydroxyl and hydroperoxyl radicals: a quantum chemistry study
Abstract
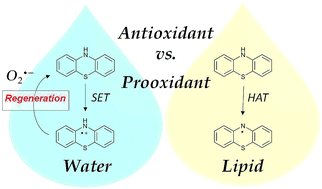
In this work, we have carried out a quantum chemistry and computational kinetics study on the reactivity of phenothiazine (PTZ) towards hydroxyl (˙OH) and hydroperoxyl (˙OOH) free radicals, in order to elucidate the antioxidant activity of phenothiazine in biological environments. We investigated three types of reaction mechanisms: (i) single electron transfer (SET), (ii) hydrogen atom transfer (HAT) and (iii) radical adduct formation (RAF). In order to mimic biological environments, we have considered both water and lipid media. We show that, in aqueous solution, PTZ acts as an excellent antioxidant while, in lipid media, it behaves as a prooxidant due to the formation of the phenothiazinyl radical that is very stable and toxic to biological systems. In addition, in water, we suggest that PTZ is able to regenerate by means of the reaction between the radical cation formed initially by electron transfer to the attacking radical, PTZ˙+, and a superoxide radical anion, O2˙−. In this process, PTZ would hence be able to scavenge two radicals per cycle (the ˙OH or ˙OOH original attacking radical, and a superoxide radical anion, O2˙−) and to form molecular oxygen O2 in situ. Finally, we show that the dication PTZ++ that has been observed experimentally in water, can be easily formed if PTZ˙+ reacts with a second ˙OH radical.
 Please wait while we load your content...
Please wait while we load your content...

