
Effect of flexible linker length in 3,4-ethylenedioxythiophene derivatives for solid state polymerization†
Chuong Tusy,
Kai Peng,
Lili Huang and
Jiangbin Xia*
College of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, China. E-mail: jbxia@whu.edu.cn; Fax: +86-27-68756707; Tel: +86-27-68756707
First published on 12th January 2015
Abstract
Previously, we made a great breakthrough in solid state polymerization (SSP) for polythiophene by the new parallel design strategy of the introduction of a one-atom linker between the 3,4-ethylenedioxythiophene (EDOT) unit. Following this strategy, several EDOT derivatives with different lengths, by simply tuning the linker atoms number, were rationally designed, synthesized and studied as potential candidates for SSP. Now the question is raised here: does the flexible linker have a limited length for successful SSP? Here, we chose EDOT-linker-EDOT as a prototype monomer model due to its facile synthetic procedure and tunable flexible linker length. We examined up to a four-atom length and also investigated different halogen substituted effects. Detailed characterizations of their corresponding polymers were carried out and crystals of all the monomers were obtained for structures analysis. Our results reveal that the successful linker length of the monomers can be up to three atoms at least because the four-atom linker monomer failed to pass SSP due to its low melting point, though it has a preferred polymerization pathway according to the theoretical prediction based on its crystal structure. Moreover, the iodo-substituted monomer is inclined to form the corresponding polymer when compared with the bromo-substituted monomer. Furthermore, it is the first time that the molecule weight information of a polymer is obtained with the observation of its dependence on the temperature involved during the SSP. Our results are very important for fully understanding the SSP process, as well as for exploring monomer structures, for obtaining further rational design of new monomers and for tuning of the corresponding polymers properties.
Introduction
The conductive polymer, which was invented in 1977, is an attractive material.1 It is an amazing functional material because of its wide applications in light-emitting devices,2,3 supercapacitors, electrochromic devices and organic photovoltaic cells.4 In addition, it can be easily tailored by rational design and synthesis. As far as its synthetic methods are concerned, they mainly include chemical oxidation polymerization, electropolymerization and photo-polymerization. Recently, the new method of solid state polymerization (SSP) has been attracting much attention due to its special features such as being solvent-free and oxidant or external applied potential free, which are necessary requirements of the traditional methods. The first polythiophene by SSP was successfully realized one decade ago;5 subsequently, several groups focused on finding novel suitable monomers and analyzing their crystal structures.6Generally, among all the monomers developed to date, the symmetrical dihalogen-substituted monomers6 and 3,4-ethylenedioxythiophene (EDOT) framework are necessary. Moreover, it is noted that all the successful dihalogen-substituted monomers are modified through the longitudinal direction of the EDOT molecule. For instance, the oxygen atoms of EDOT were replaced by sulfur or selenium,6a or the sulfur atom in the thiophen ring was replaced by selenium.6d In addition, the modification on EDOT, whether based on the EDOT framework or its derivative, is tedious involving a multi-step synthesis. Due to such restriction based on the present monomer design strategy, recently, we successfully designed a monomer along the parallel direction of EDOT by introducing a flexible linker between the EDOT units, and we obtained the corresponding conductive polymers by SSP.7
Moreover, the widely investigated and million-ton scale of non-conjugated polyester amides was obtained by SSP. Generally, these polymers were formed from polyfunctional monomers through various organic chemistry reactions, resulting in repeating units of esters, amides, and ethers. Previous studies reveal that sometimes crystal structures, especially crystals size, have a strong effect on their SSP process, and collision and steric factors may contribute to the polymerization reaction as well.8a–b Scientists have paid considerable attention on SSP through exploring new reactions and applications.8c–e
It should be noted that though the introduction of a non-conjugated linker results in the generation of non-conjugated polymers, it demonstrates the possible limitation of the monomer size for SSP, irrespective of it being a conjugated or a non-conjugated monomer. In addition, compared with the well-investigated polyester,8f few studies concerning thiophene containing non-conjugated polymers through SSP have been reported.
Here, we further examined the flexible linker length effect among the EDOT derivatives and checked their SSP behavior. This study not only reveals fundamental information on SSP, but also broadens the monomer design concept and we obtained novel polymers as well.
Experimental
Materials
Chemicals were purchased from Wuhan Shenshi Chemicals Co., Ltd. and were used without further purification unless otherwise noted. EDOT was purchased from J & K. 5-Bromo-3,4-ethylenedioxythiophene9 was synthesized according to our previous report. 5-Iodo-2,3-dihydro-thieno[3,4-b][1,4]dioxine10 was synthesized similar to 5-bromo-3,4-ethylenedioxythiophene except that 5,7-diiodo-3,4-ethylenedioxythiophene was used during the reduction by n-butyllithium.Monomer synthesis and solid state polymerization
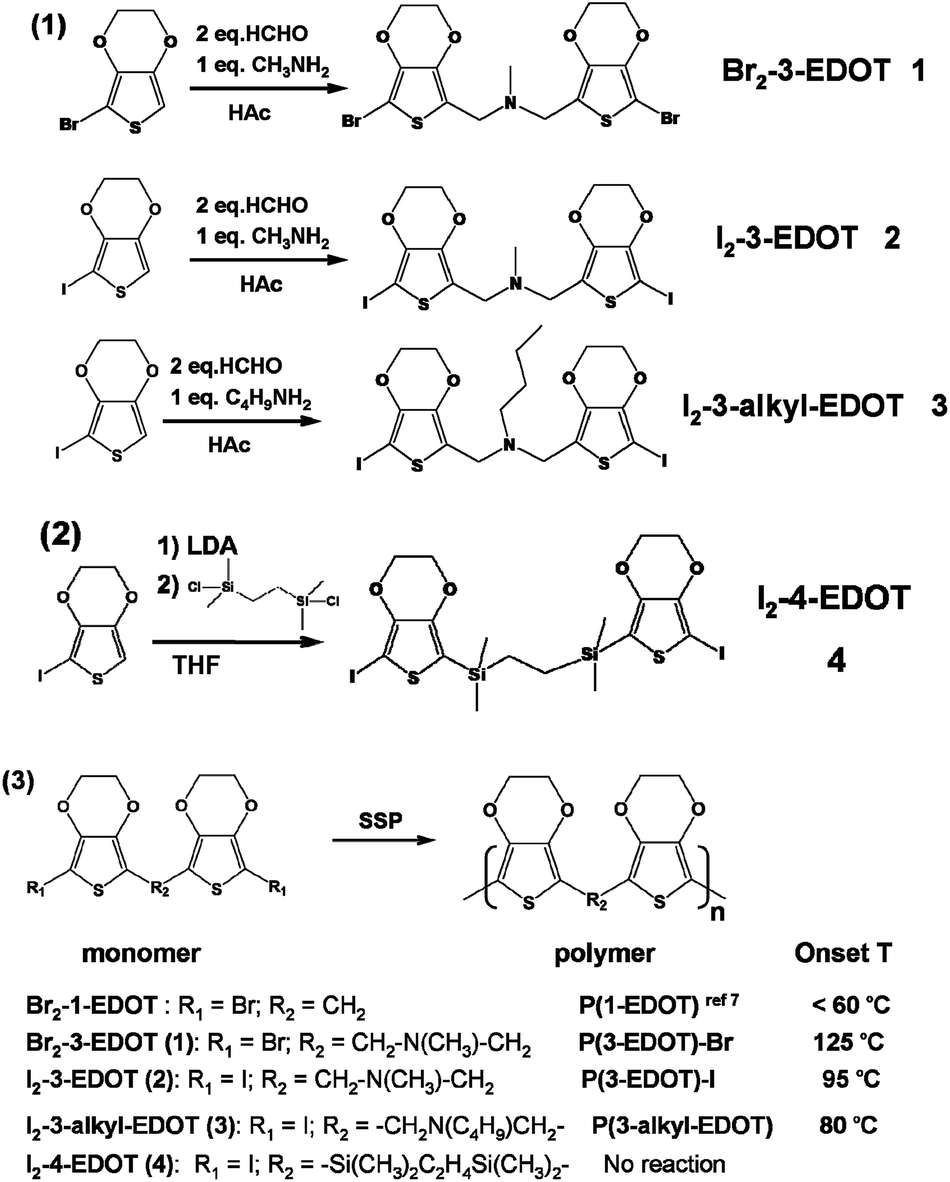 | ||
| Scheme 1 Synthesis of the monomers and their corresponding polymers. | ||
Bis-(7-iodo-2,3-dihydro-thieno[3,4-b][1,4]dioxin-5-ylmethyl)-methyl-amine (Br2-3-EDOT, 1). Yellow-brown 37%, 1H NMR: δ (CDCl3) 4.25 (m, 4H); 4.20 (m, 4H); 3.59 (s, 4H), 2.28 (s, 3H). 13C NMR: δ (CDCl3) 139.7, 138.7, 115.3, 85.8, 65.4, 64.8, 51.2, 42.0. Calcd for C15H15I2NO4S2: C, 36.23%; H, 3.04%; N, 2.82%. Found: C, 30.50%; H, 2.56%; N, 2.56%.
Bis-(7-iodo-2,3-dihydro-thieno[3,4-b][1,4]dioxin-5-ylmethyl)-methyl-amine (I2-3-EDOT, 2). Yellow-brown (35%), 1H NMR: δ (CDCl3) 4.25 (m, 4H); 4.17 (m, 4H); 3.62 (s, 4H); 2.28 (s, 3H). 13C NMR: δ (CDCl3) 143.8, 138.6, 120.2, 65.4, 64.7, 51.4, 47.7, 41.9. ESI-MS: calcd, 590.8; found (M + 1)+, 591.6. Calcd for C15H15I2NO4S2: C, 30.47%; H, 2.56%; N, 2.37%. Found: C, 30.50%; H, 2.56%; N, 2.56%.
Butyl-bis-(7-iodo-2,3-dihydro-thieno[3,4-b][1,4]dioxin-5-ylmethyl)-amine (I2-3-butyl-EDOT, 3). Yellow solid (40%), 1H NMR: δ (CDCl3) 4.23–4.37 (m, 4H); 4.13–4.18 (m, 4H); 3.67 (s, 4H); 2.42–2.49 (t, 2H); 1.44–1.51 (m, 2H); 1.25–1.36 (m, 2H); 0.85–0.92 (t, 3H). 13C NMR: δ (CDCl3) 143.9, 138.4, 121.5, 65.6, 64.8, 52.9, 48.8, 47.6, 29.6, 20.7, 14.4. Calcd for C18H21I2NO4S2: C, 34.14%; H, 3.34%, N, 2.21%. Found: C, 35.72%; H, 3.31%; N, 2.66%.
1,2-Bis-(7-iodo-2,3-dihydro-thieno[3,4-b][1,4]dioxin-5-yl)-bis-dimethylsilanyl-methane (I2-4-EDOT, 4). Monomer 4 (I2-4-EDOT) was synthesized by the modification of a previous method.7,12 Anhydrous THF (30 ml) was slowly added dropwise to a solution of lithium diisopropylamide (10 mmol), and the temperature was maintained around −15 °C. The reaction mixture was cooled to −60 °C, and a solution of 5-iodo-3,4-ethylenedioxythiophene (2.21 g, 10 mmol) in anhydrous THF (10 ml) was slowly added dropwise, and the temperature was maintained around −50 °C. Then, the reaction mixture was warmed up to −20 °C and it was stirred at this temperature for 15 min. Subsequently, the reaction mixture was cooled to −78 °C and dichlorodimethylsilane (0.65 g, 5 mmol) was added, and then the cooling bath was removed and the temperature was increased to room temperature. After the completion of the reaction, the mixture was poured into a mixture of ice water (50 ml) containing 2 ml 1 M HCl and freshly distilled ether (100 ml). The organic layer was washed with water and dried with Na2SO4. Purification by column chromatography (silica gel, light petroleum–CH2Cl2, 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) afforded the colorless crystal 4 (50%). 1H NMR: δ (CDCl3) 4.20–4.30 (m, 4H); 0.67 (s, 4H); 0.24 (s, 12H). 13C NMR: δ (CDCl3) 146.5, 145.0, 117.0, 65.2, 64.6, 61.8, 54.4, 8.1, −2.9. Calcd for C14H14Br2O4S2Si: C, 33.75%; H, 2.83%. Found: C, 32.95%; H, 2.96%.
:1) afforded the colorless crystal 4 (50%). 1H NMR: δ (CDCl3) 4.20–4.30 (m, 4H); 0.67 (s, 4H); 0.24 (s, 12H). 13C NMR: δ (CDCl3) 146.5, 145.0, 117.0, 65.2, 64.6, 61.8, 54.4, 8.1, −2.9. Calcd for C14H14Br2O4S2Si: C, 33.75%; H, 2.83%. Found: C, 32.95%; H, 2.96%.
General solid state polymerization
The SSP procedure was carried out according to a previous method.5,7 In a closed 5 ml vial, the brominated and iodinated compounds (300 mg) were incubated at 60 °C and 130 °C, respectively, for 2–72 h. The grounded polymers were additionally dried in vacuum at 80 °C overnight, and then stirred with hydrazine hydrate (50% aqueous solution, in CH3OH) overnight, filtered, and washed with CH3OH, and vacuum drying afforded a hydrazine-treated polymers.Results and discussion
Synthesis of monomers and solid state polymerization
All the designed monomers were synthesized as shown in Scheme 1. Due to the difficulty of the final bromination step, monobromo substituted EDOT was directly synthesized as the starting material. Monomers 1–3 were obtained through the Mannich reaction to link the thiophene moiety of 5-bromo or iodo substituted 3,4-ethylenedioxythiophene. Compound 4 was obtained by the reaction of mono-iodo substituted EDOT with 1,2-bis-dimethylsilanyl-ethane with the help of a Li-reagent, LDA.Finally, these resulting dihalogen-substituted monomers were polymerized through a self-polycondensation process with the formation of halogen vapor under SSP. According to the SSP results, except for monomer 4, other monomers can successfully form their corresponding polymers, indicating that linker distance plays a key role in their SSP behavior. Previously, we introduced a one-atom C, Si or P linker between EDOT units and got a great success in SSP.7 Therefore, we can draw the conclusion that a flexible linker with a single bond and up to three atoms has an appropriate length for a successful SSP. Though we failed to obtain EDOT monomers with a two-atom-length spacer, it can be expected that such structure could be a successful candidate for SSP. Therefore, a flexible linker along the parallel direction proves to be an excellent design strategy and the maximum length would be up to three atoms according to our results. A detailed discussion is done in the following crystal analysis section. To establish the relationship between structure and SSP behavior, we define the onset temperature of SSP (Tonset) as the temperature that results in 10% of conversion efficiency in 24 hours and all the Tonset are listed in Scheme 1.
It is interesting to note that after the introduction of the long butyl chain, monomer 3 requires a lower temperature, about 15 °C, to complete SSP when compared with monomer 2, which needs 95 °C, while the monomer 1 needs the highest temperature of 125 °C. In addition, previously Perepichka's group observed that the fixed 5-member ring monomers were more stable than the free two methyl group molecules attached on ethylene 2,3-dihydro-thieno[3,4-b][1,4]dioxine.6c All these facts reveal that the substitution groups, whether flexible or rigid, have a great effect on their SSP behavior, especially on the Tonset, which encouraged us to further explore much more acutely controllable and fine tunable monomers for SSP.
XRD patterns of monomers and respective polymers
As shown in Fig. 1, the monomer of I2-3-EDOT shows typical sharp peaks (2θ = 12.2°, 22.0°, 23.2°, 24.8°, 26.0° and 32.5°) in the rage of 5–60°, whereas Br2-3-EDOT has sharp peaks (2θ = 12.4°, 13.7°, 17.8°, 18.3°, 22.3°, 23.2°, 23.7°, and 25.3°) as well, which are characteristic features for crystal compounds. After butyl was attached on the nitrogen, I2-3-alkyl-EDOT changes its original structure with peaks at 13.8°, 15.8°, 19.5°, 22.0°, 26.7° and 29.3°, as indicated in its XRD pattern. Moreover, all these peak values are considerably consistent with their simulated results based on the crystal structure data. After polymerization, all the polymers are in amorphous phase due to the halogen release, which results in the collapse of the crystals.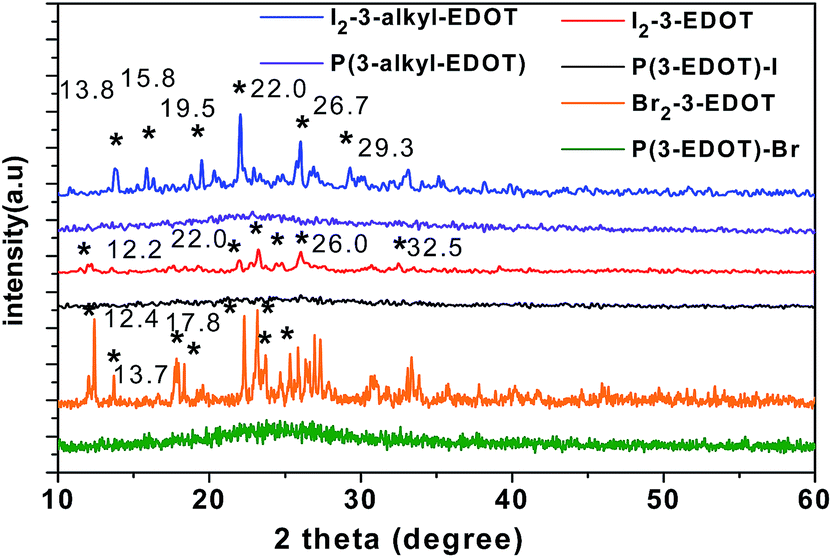 | ||
| Fig. 1 XRD spectra of monomers and their corresponding polymers. | ||
Absorption of polymers by SSP and hydrazine treated polymers
Fig. 2 shows the absorbance spectra of P(3-EDOT)-Br, P(3-EDOT)-I, P(3-alkyl-EDOT) and their corresponding hydrazine treated polymers. It is obvious that these polymers obtained through SSP do not show typical broad peaks in the near-IR region, indicating that these polymer were in the low doped (oxidized) state.14–16 In addition, after treating with hydrazine, there were no distinct peaks in the region of 540–650 nm, which are frequently observed in polythiophene and ascribed to the π–π* electronic transition of dedoped (reduced) polymers.15,16 Previously,7 we observed such typical peaks in the polymers obtained through SSP based one-atom linker EDOT monomers. Therefore, we attribute such special phenomena to the poor conjugation of the polymer due to the long three-atom linker existing in the polymer matrix. Thus, due to the introduction of a non-conjugated linker, the obtained polymers are non-conjugated without the typical p-type feature peaks in their absorbtion spectra under SSP. Thus, hydrazine treatment has little effect on the absorption spectra of films as well.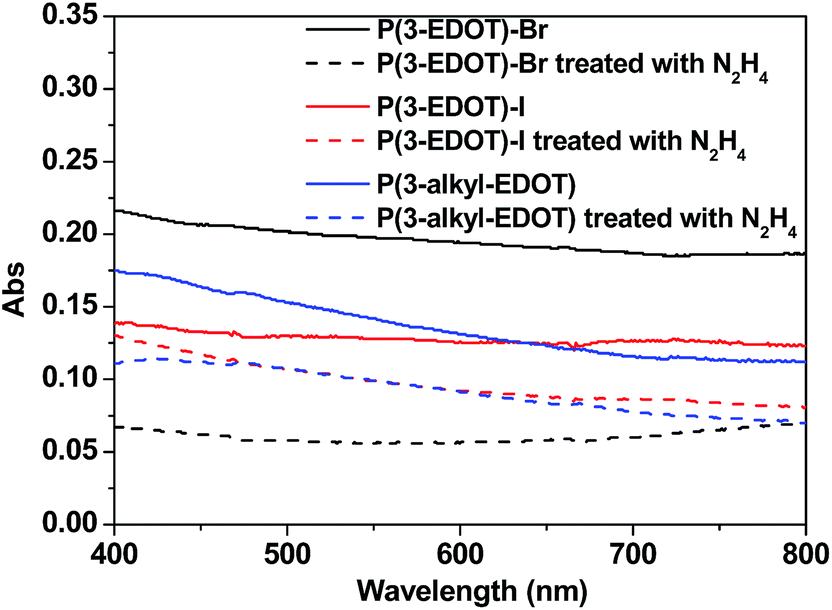 | ||
| Fig. 2 Absorbance spectra of the polymers and the hydrazine treated polymers (freshly made). | ||
FTIR spectroscopy
Structural information was obtained using Fourier transform infrared (FTIR) spectrometry, as shown in Fig. 3. All the polymers have peaks around 1480, 1439, 1340, and 1200 cm−1, which originated from the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C in the thiophene ring.17,18 As we noted, all the polymers shows distinct strong sharp peak at 2920 and 2850 cm−1, which are assigned to the CH3 stretching modes.19 Due to the existence of C–N vibrations, a peak at 1080 cm−1 was observed.20
C and C–C in the thiophene ring.17,18 As we noted, all the polymers shows distinct strong sharp peak at 2920 and 2850 cm−1, which are assigned to the CH3 stretching modes.19 Due to the existence of C–N vibrations, a peak at 1080 cm−1 was observed.20
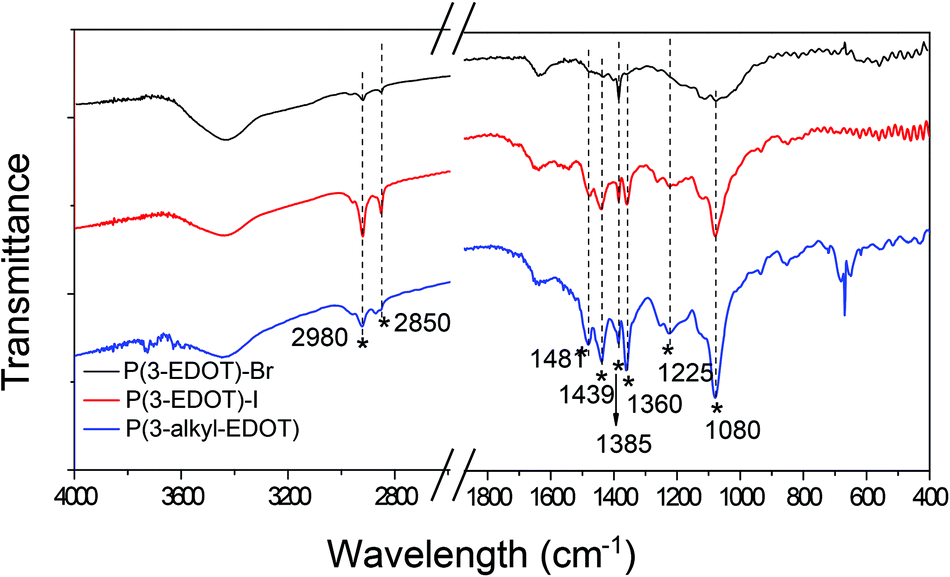 | ||
| Fig. 3 FTIR spectra for the polymers. | ||
Photo-image and SEM images
After SSP at the desired temperature overnight, the yellow-brown or yellow crystals of the monomers changed to a sky-blue or black colored powder, as shown in Fig. 4a. Fig. 4b–g show the SEM surface morphologies of the drop-casted monomers films and their respective polymers on an FTO substrate. It is obvious that there is a great change in the morphology after SSP. Except for Br2-3-EDOT, most of the monomers show typical aggregation of micrometer-sized irregular-shape sheets, while most of the polymers show similar shapes with the typical layered structure. It is noticed that in the case of P(3-alkyl-EDOT), it shows a different morphology with spheres or ellipsoids with sizes ranging from 0.3–2 μm, and spheres with sizes as high as 10 μm were also observed. Such characteristic features should be attributed to its lower Tg because of the observation of large volume shrinkage under the higher SSP temperature. | ||
| Fig. 4 (a) Digital imagines of crystals of monomers and their respective polymers. (b) Br2-3-EDOT, (c) P(3-EDOT)-Br, (d) I2-3-EDOT, (e) P(3-EDOT)-I, (f) I2-3-alkyl-EDOT and (g) P(3-alkyl-EDOT). Different scale views are shown as insets in (b), (c) and (g). | ||
Crystallographic X-ray analysis
Single crystals of the three successful monomers (1, 2 and 3) and the failure sample 4 were obtained and investigated by X-ray analysis (shown in Fig. 5–9) for further understanding the structural requirements and SSP polymerization pathway. All the parameters of these crystals are summarized in Table S1 in the ESI.† As we can see, 1 and 2 share the same space group and they almost have the same crystal parameter values, namely, a, b, c. After the methyl on the nitrogen of 2 was replaced by the butyl of 3, there were great changes from the space group of P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (triclinic) to the space group of P
(triclinic) to the space group of P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c (tetragonal) in the crystal, which shows considerably different structural information, as summarized in Table S1 (see ESI†). In addition, the number of molecules contained in a unit cell increases from two to eight. In the case of I2-4-EDOT (4), it belongs to monoclinic space group of P21/c. The monomer structures of 1–4 are shown in Fig. 5. The distance between S–S is 5.112 and 5.136 Å for Br2-3-EDOT and I2-3-EDOT, respectively, while the introduction of the alkyl chain results in the shortest distance of 3.781 Å. However, in the case of I2-4-EDOT, the –Si–(CH2)2–Si– linker greatly enhances the S–S distance to as high as 7.295 Å. Here, we discuss the crystal structures of monomers and try to establish the relationship between their structural information and their SSP behavior.
21c (tetragonal) in the crystal, which shows considerably different structural information, as summarized in Table S1 (see ESI†). In addition, the number of molecules contained in a unit cell increases from two to eight. In the case of I2-4-EDOT (4), it belongs to monoclinic space group of P21/c. The monomer structures of 1–4 are shown in Fig. 5. The distance between S–S is 5.112 and 5.136 Å for Br2-3-EDOT and I2-3-EDOT, respectively, while the introduction of the alkyl chain results in the shortest distance of 3.781 Å. However, in the case of I2-4-EDOT, the –Si–(CH2)2–Si– linker greatly enhances the S–S distance to as high as 7.295 Å. Here, we discuss the crystal structures of monomers and try to establish the relationship between their structural information and their SSP behavior.
 | ||
| Fig. 5 Crystal structure of (a) Br2-3-EDOT, (b) I2-3-EDOT, (c) I2-3-alkyl-EDOT and (d) I2-4-EDOT. | ||
 | ||
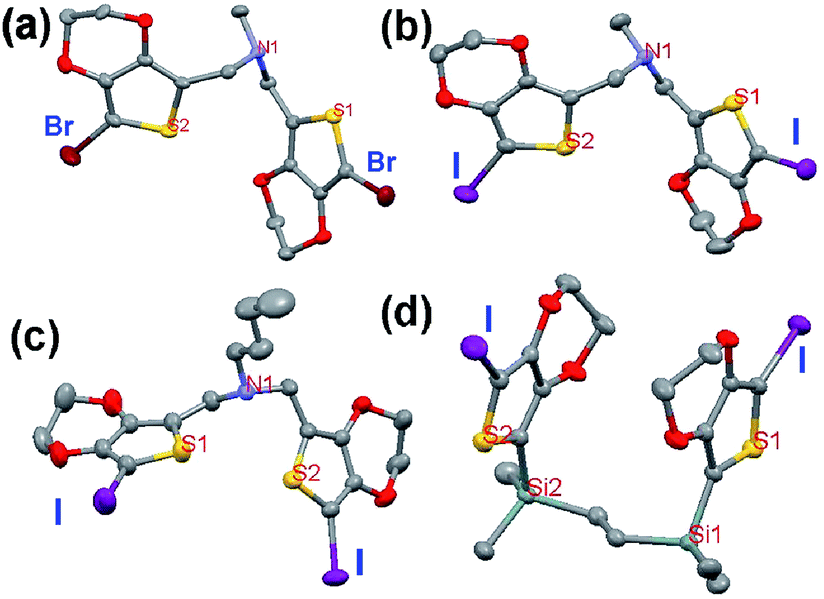
| Fig. 6 Single-crystal X-ray structure of compound Br2-3-EDOT (1). (a) View of the halogen distances, (b) view of C/C contact distances, (c) crystal packing viewed along the a-axis, C1/C1 and C1/C15 distance between each third molecule and proposed polymerization pathways. The numbers indicate the corresponding distances in Å. Hydrogen atoms are omitted for clarity: Br, red; S, yellow; N, light blue and C, gray. | ||
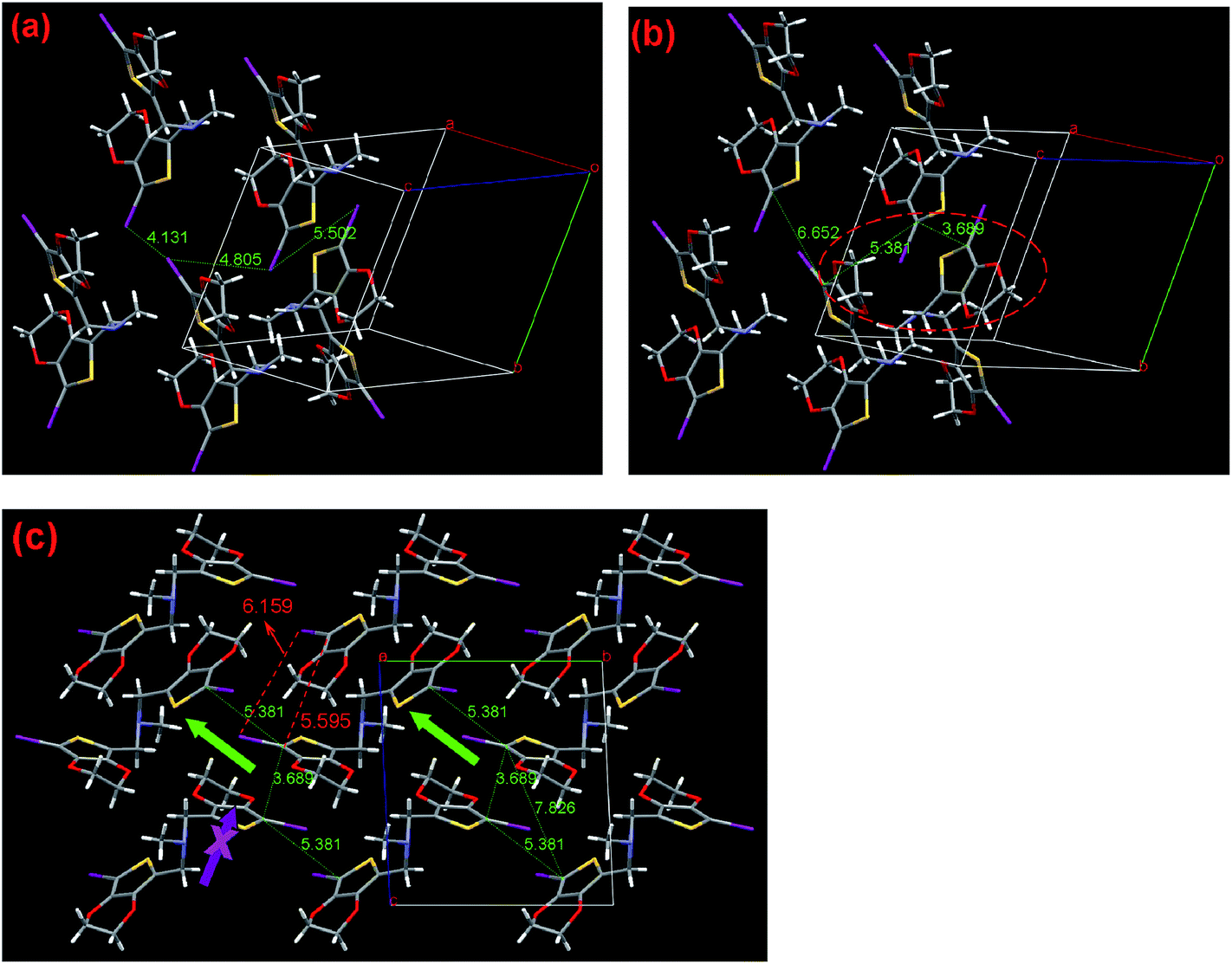 | ||
| Fig. 7 Single-crystal X-ray structure of compound I2-3-EDOT (2). (a) View of the halogen distances, (b) view of C/C distances, (c) crystal packing viewed along the a-axis, C1/C13 distance between each third molecule and proposed polymerization pathways. The numbers indicate the corresponding distances in Å. I, purple; S, yellow; N, light blue and C, gray. | ||
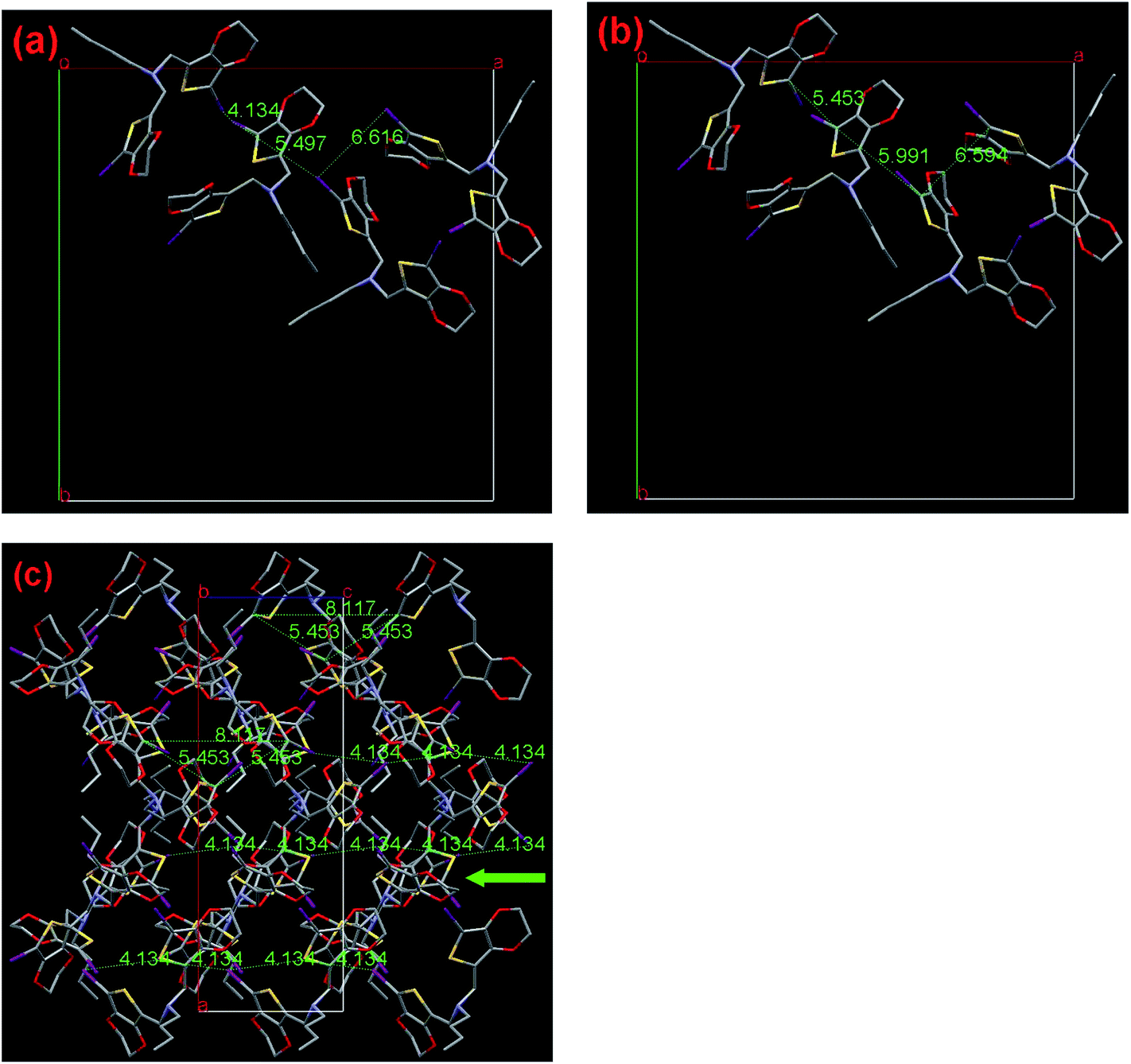 | ||
| Fig. 8 Single-crystal X-ray structure of compound I2-3-alkyl-EDOT (3). (a) View of the halogen distances, (b) view of C/C distances, (c) crystal packing viewed along the a-axis, C1/C13 distance between each third molecule and proposed polymerization pathways. The numbers indicate the corresponding distances in Å. Hydrogen atoms are omitted for clarity: I, purple; S, yellow; N, light blue and C, gray. | ||
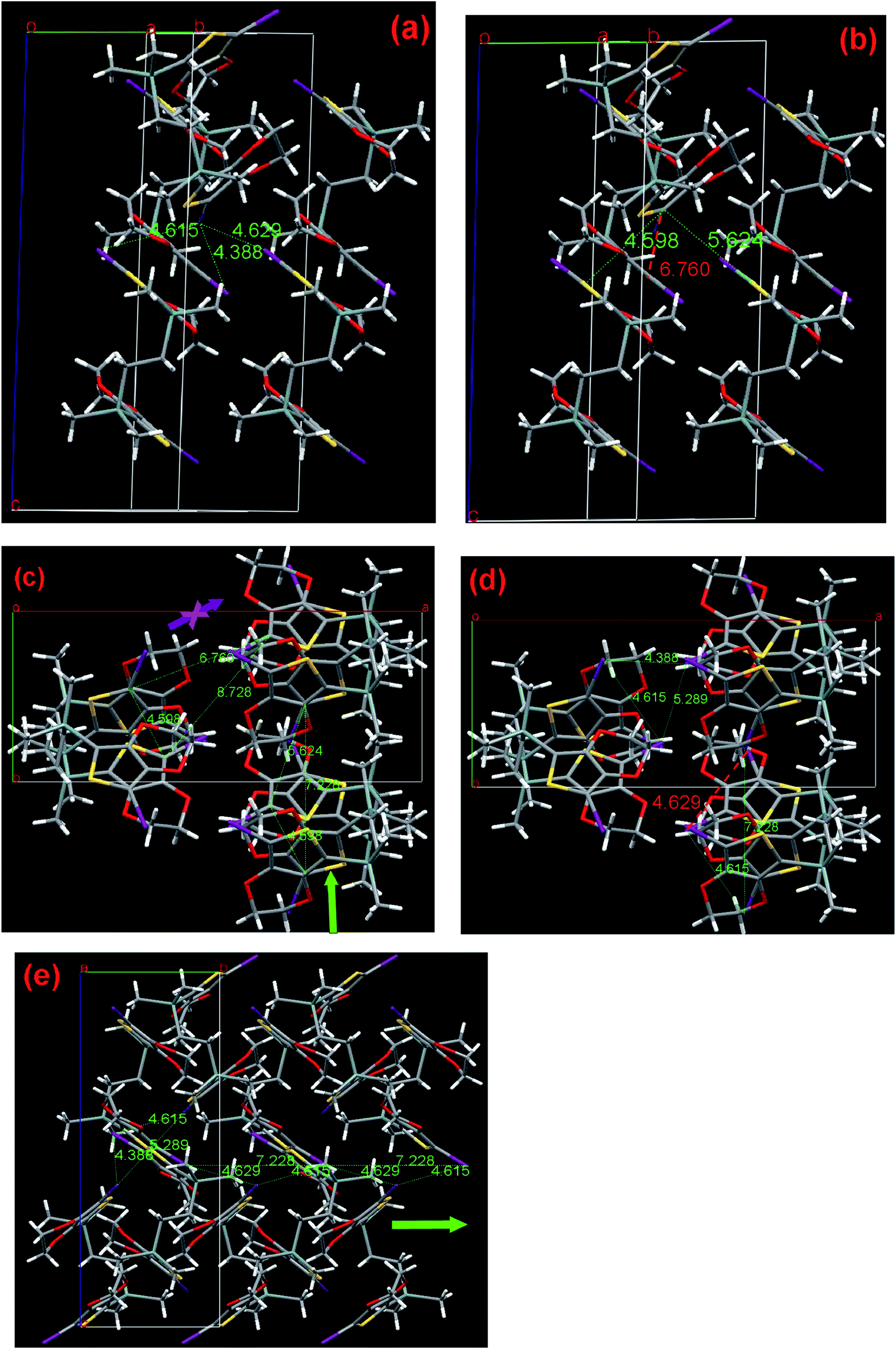 | ||
| Fig. 9 Single-crystal X-ray structure of compound I2-4-EDOT (3). (a) View of the halogen distances, (b) view of C/C distances, (c) crystal packing viewed along the c-axis, C1/C1, C18/18 distance between each third molecule and proposed polymerization pathways. (d) Corresponding Hal/Hal distance based on the C/C contact of (c). (e) Crystal packing viewed along the a-axis. The numbers indicate the corresponding distances in Å. Hydrogen atoms are omitted for clarity: I, purple; S, yellow; Si and C, gray. | ||
As shown in Fig. 6a and b, 1 has the nearest halogen distance of 4.067 Å with the C1–C15 contact of 6.278 Å, followed by the second shortest distance of 4.678 Å with the C1–C15 contact of 5.126 Å and then having the third shortest distance of 5.369 Å with the shortest C1–C1 contact of 3.568 Å. Previous studies6,7a showed that SSP pathways usually involve a C/C triangle unit composed of three monomer molecules in the chain. Here, the new polymerization mode comprising of four monomer molecules appeared, as shown in Fig. 6c. In such case, the polymerization process should be demonstrated in the following way: first 1 forms a trimer with the construction of a triangle through C/C contact in a regular mode, and then the trimer attacks the fourth monomer to complete the repeatable four sub-units for the further construction of its polymer matrix, as demonstrated with the C15–C1–C1–C15 pathway. In this case, the fact that the initial trigger of C15/C1 has a short halogen distance, i.e., an effective halogen distance of 4.678 Å with 26.4% longer than double van der Waals radius of halogen (2rw), can well explain that 1 has SSP success with its SSP onset temperature (Tonset) of 125 °C.
Therefore, there are two possible modes of C15–C1–C1–C15 (Fig. 6c) and a reverse one and all of their Hal/Hal and C/C distances are suitable for SSP. It is obvious that the former mode is the preferred polymerization pathway because the initial trigger C1/C15 has a shorter halogen distance of 4.678 Å while the latter has that of 5.369 Å.
After bromo was replaced by iodo in monomer 2, its crystal structure basic parameters are almost the same as monomer 1. However, there was still some change in detail. As shown in Fig. 7a and b, 2 has nearest halogen distance of 4.131 Å with C1–C13 contact of 6.652 Å, followed by the second shortest distance of 5.502 Å with C1–C1 having the shortest contact of 3.689 Å. It is obvious that though the former mode has the nearest halogen distance, its C1/C13 distance (6.652 Å) is too long to form the corresponding polymer. As shown in Fig. 7c in a view along the a-axis, there are two possible pathways for the polymerization. One pathway, as denoted by the blue arrow, is that the C1–C13 contact distance is 5.381 Å with the Hal/Hal distance of 4.805 Å, while the neighbor C1–C1 contact of 3.689 Å and Hal/Hal of 5.502 Å are found in the crystal. Moreover, the C1/C13 distance between each third molecule in the chain (7.826 Å) is almost the same as the corresponding distance in regular polythiophene (7.85 Å),21 which is shorter than those having one atom-spacer derivatives, as previously reported by us.7
Another possible polymerization direction (denoted with the purple arrow) is as follows. The C1–C13 contact distance is 5.381 Å, with the Hal/Hal distance of 4.805 Å, while the neighbor C1–C1 contact of 5.595 Å and very long Hal/Hal of 6.159 Å are found in the crystal. Moreover, the C1/C13 distance between each third molecule in the chain is 9.101 Å. Thus, both modes share the first initial dimer formation procedure with the effective Hal/Hal distance of 4.805 Å. Therefore, we assume that the former pathway might be a more favorable choice because of the shorter Hal/Hal distance involved. In addition, we noted that the effective Hal/Hal distance (4.805 Å) is considerably longer (20.1%) than the 2rw of iodine (4.0 Å). This result matches our expectation that the flexible three-atom-spacer must have a long halogen distance to guarantee SSP success.
As shown in Fig. 8a and b, 3 has the nearest halogen distance of 4.134 Å with the C12–C12 contact of 5.453 Å, followed by the second shortest distance of 5.497 Å with C6–C12 having the shortest contact of 5.991 Å and the third shortest distance of 6.616 Å with C6–C6 having the shortest contact of 6.594 Å are found in the crystal. Moreover, the shortest C12/C12 contacts of 5.453 Å, observed between the EDOT units with a dihedral angle of 83.35°, is shorter than the corresponding distance in 2. These contacts form four one-dimensional chains in one crystal unit cell, which is considerably different to that observed for 2, but running along the crystallographic c-axis. In this case, the C12/C12 distance (8.117 Å) between each third molecule in the chain is only 3% longer than the corresponding distance in the traditional polythiophene chain (7.85 Å), which is favorable for SSP along this direction. Due to the fact that the other pathway has a too long distance to construct polythiophene, the sole and reasonable polymerization pathway is along the c-axis, as clearly shown in Fig. 8c. According to our previous study of one-atom linkers with V-shape monomers, including monomer 1 and 2 as discussed above, 3 demonstrates a considerably different polymerization pathway, involving the same distance of 4.134 Å, which is frequently observed in the investigated small EDOT derivatives in the SSP field.6
In addition, considering that 3 is less stable than 2, as mentioned in the SSP behavior section, one may deduce that though the bulkier butyl chain would occupy bigger room in the crystal packing and would be unfavorable for SSP, the butyl is considerably more flexible, which helps the V shaped molecule rotate more freely than the methyl containing monomer. This is the simple qualitative explanation. In fact, 3 has a shorter effective halogen distance of 4.134 Å, while 2 has as high as 4.805 Å, which are longer than 2rd by 3.4% and 20.1%, respectively. This is a solid evidence to explain why 3 more easily undergoes SSP compared with 2. According to these facts, we can conclude that the replacement of the methyl with butyl group greatly changes the crystal structure as well as SSP behavior of the monomer. Therefore, this finding may offer considerable opportunity to further rationally and accurately control the SSP behavior of monomer through a simple modification of the steric structure of the linker.
After a long flexible four-atom-chain was introduced, I2-4-EDOT demonstrates a folded molecule with a torsion angle of 50.31° and the two EDOT subunits are almost parallel with each other with their C(EDOT)–Si–CH2 angles of 107.65° and 105.65°, respectively. As shown in Fig. 9a and b, 4 has the shortest, second and third halogen distance of 4.388, 4.615, and 4.629 Å with the corresponding C1–C18 contact of 6.760, 4.598 and 5.624 Å, respectively. Similar to I2-3-EDOT (2), there are two possible polymerization pathways (shown in Fig. 9c), which are demonstrated by two triangles of C/C distance and corresponding triangles of Hal/Hal distance. In these two cases, it is noted that the C/C distance between each third molecule in the chain (8.728 Å for C1/C1, 7.228 Å for C18/C18) are suitable and the Hal/Hal distances (4.388, 4.615; 4.615, 4.629 Å) are short enough as well, which may freely construct the corresponding polymers. However, considering the shortest Hal/Hal distance of 4.386 Å with the extraordinary long C1/C18 contact of 6.760 Å, this is the unfavorable polymerization pathway, as denoted by the purple arrow, and the preferred one was presented as well (blue arrow sign) in Fig. 9c. However, the SSP failure of I2-4-EDOT contradicts the theoretical prediction. Compared with the Tonset of I2-3-EDOT (95 °C), we cannot expect a lower Tonset of I2-4-EDOT because they do not share the same frame structure due to different linker lengths. We attribute such interesting observation to its relatively low melting point of 115–116 °C. In this case, its crystal structure collapses before it reaches Tonset. Such special result reveals that the crystal structure analysis is a powerful technique to guide monomer design and predict its SSP behavior; however, sometimes the prediction would be invalid in the case of low melting point monomers.
Through the careful discussion above, Table 1 summarizes all important distances derived from the crystal structures. As we can see, the effective Hal/Hal distances for Br2-3-EDOT, I2-3-EDOT and I2-3-alkyl-EDOT are longer than 28.7%, 20.1% and 3.4% of their 2rw, respectively, and all of them succeed in SSP. Though I2-4-EDOT is 15.4% longer than the 2rw of iodine, it fails to pass the SSP test due to its lower melting point. We are trying to establish the relationship between effective Hal/Hal distances with onset SSP temperature by collecting numerous crystal data. However, it needs up to one hundred samples because the variation of the main structure and substitution groups has a big effect on Tonset.
| Molecules parameters | Br2-3-EDOT | I2-3-EDOT | I2-3-alkyl-EDOT | I2-4-EDOT |
|---|---|---|---|---|
| a Effective Hal/Hal distance; 2rw: double van der Waals radius of halogen. | ||||
| Shortest Hal/Hal distance | 4.067 | 4.131 | 4.134a | 4.388 |
| 2nd shortest Hal/Hal distance | 4.678a | 4.805a | 5.497 | 4.615a |
| 3rd shortest Hal/Hal distance | 5.369 | 5.502 | 6.616 | 4.629 |
| C–C contact shortest distance | 3.568 | 3.689 | 5.453 | 4.589 |
| C–C contact 2nd shortest distance | 5.126 | 5.381 | 5.991 | 5.624 |
| C/C distance between each third molecule | 8.396 | 7.826 | 8.117 | 7.228 |
| 2rw | 3.7 | 4.0 | 4.0 | 4.0 |
| Longer than 2rwa | 26.4% | 20.1% | 3.4% | 15.4% |
| Onset of temperature | 125 °C | 95 °C | 80 °C | — |
Molecular weight and thermal stability TGA analysis
Because of the insolubility of the other two polymers, the soluble P(3-alkyl-EDOT) was chosen as a typical model compound for further characterization, especially the concerning molecular weight and its GPC profiles are presented in Fig. S10 (ESI†). Previously, MALDI-TOF mass spectrometry revealed that poly(2,3-dihydro-thieno[3,4-b][1,4]dithiine) obtained through SSP contains up to 5–14 monomer units, and the elemental analysis of the dedoped polymer indicates an average of six repeat units per chain.6a Here, we get the typical polymer molecular weight information of P(3-alkyl-EDOT) through GPC. The Mn of 4.40 kg mol−1 with PDI of 2.19 indicates that the P(3-alkyl-EDOT) has a repeat unit of 10, which is consistent with the results described above. Moreover, the temperature dependence with Mn and PDI is summarized in Table 2. As we can see, a higher temperature results in a slightly lower molecular weight and the extension of SSP time does not have great influence on Mn. The thermal stability of the polymers was investigated with thermogravimetric analysis (TGA) (Fig. S11†). The 5% weight-loss temperatures for P(3-EDOT)-Br, P(3-EDOT)-I and P(3-alkyl-EDOT) were 193, 185 and 248 °C, respectively, indicating that the introduction of the alkyl chain would greatly enhance the thermal stability. In addition, it appears that P(3-EDOT)-I shows less stability compared with P(3-EDOT)-Br. We attribute this to its lower stability with the high halogen content in the polymer matrix, which results in quick halogen release under thermal treatment.| Temperature | Yield (%) | Retention time | Mna (kg mol−1) | DPb | PDIa |
|---|---|---|---|---|---|
| a Molecular weights and PDI were determined by GPC with respect to polystyrene standards.b Degree of polymerization (DP) was calculated based on the Mn values determined by GPC. | |||||
| 80 °C | >90 | 23.55 | 4.40 | 11 | 2.19 |
| 90 °C | >90 | 24.4 | 3.83 | 10 | 2.16 |
| 100 °C | >90 | 24.7 | 3.64 | 9 | 1.94 |
| 80 °C (3 days) | >90 | 24.1 | 4.11 | 10 | 2.20 |
Conclusion
In this work, several types of new EDOT derivatives containing flexible linkers of different lengths were synthesized and investigated under SSP. Among these rationally designed monomers, the iodo-substituted monomer with flexible three-atom linkers has a similar crystal structure and easily undergoes SSP when compared with the bromo-substituted monomer. Moreover, in the case of the four-atom linker length, though it obtained SSP success theoretically according to its crystal structure analysis, it cannot be polymerized because of its low melting point, indicating that at present we did not find the length limit for the flexible chain linker. Combined with the crystal structure data, these observations and evidence may not only help to gain a deeper understanding of SSP but also enlarge the thiophene-based SSP database. Furthermore, the substitution on the flexible linker moiety also plays an important role in the fine-tuning of the SSP behavior of the monomers and the functional properties of their corresponding polymers. In addition, more investigations are underway in our lab. In conclusion, the linker length, linker structure and sometimes melting point of a monomer are key factors to determine its SSP behavior.Acknowledgements
This work was supported by the Natural Science Foundation of China (21371138).Notes and references
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578 RSC.
- Q. PEi, Y. Yang, G. Yu, C. Zhang and A. J. Heeger, J. Am. Chem. Soc., 1996, 118, 3922 CrossRef CAS.
- H. L. Wang, F. Huang, A. G. MacDiarmid, Y. Z. Wang, D. D. Gebler and A. J. Epstein, Synth. Met., 1996, 80, 97 CrossRef CAS.
- (a) J. H. Schon, Z. Bao, C. Kloc and B. Batlogg, Nature, 2000, 406, 702 CrossRef CAS PubMed; (b) J. Xia, N. Masaki, K. Jiang and S. Yanagida, J. Mater. Chem., 2007, 17, 2845 RSC; (c) J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258 CrossRef CAS PubMed; (d) J. Xia, L. Chen and S. Yanagida, J. Mater. Chem., 2011, 21, 4644 RSC; (e) L. Chen, J. Jin, X. Shu and J. Xia, J. Power Sources, 2014, 248, 1234 CrossRef CAS PubMed.
- H. Meng, D. F. Perepichka, M. Bendikov, F. Wudl, G. Z. Pan, W. Yu, W. Dong and S. Brown, J. Am. Chem. Soc., 2003, 125, 15151 CrossRef CAS PubMed.
- (a) H. J. Spencer, R. Berridge, D. J. Crouch, S. P. Wright, M. Giles, I. McCulloch, S. J. Coles, M. B. Hursthouse and P. J. Skabara, J. Mater. Chem., 2003, 13, 2075 RSC; (b) A. Patra, Y. H. Wijsboom, S. S. Zade, M. Li, Y. Sheynin, G. Leitus and M. Bendikov, J. Am. Chem. Soc., 2008, 130, 6734 CrossRef CAS PubMed; (c) M. Lepeltier, J. Hiltz, T. Lockwood, F. Bélanger-Gariépy and D. F. Perepichka, J. Mater. Chem., 2009, 19, 5167 RSC; (d) A. Patra, Y. H. Wijsboom, G. Leitus and M. Bendikov, Chem. Mater., 2011, 23, 896 CrossRef CAS; (e) S. Chen, J. Xu, B. Lu, X. Duan and F. Kong, Adv. Mater. Res., 2011, 239–242, 924 CAS.
- (a) C. Tusy, L. Huang, J. Jin and J. Xia, RSC Adv., 2014, 4, 8011 RSC; (b) C. Tusy, L. Huang, K. Peng and J. Xia, RSC Adv., 2014, 4, 29032 RSC.
- (a) T. Fadner and H. Morawetz, J. Polym. Sci., 1960, 45, 475 CrossRef CAS; (b) C. Chen and D. Grabar, J. Polym. Sci., 1964, 4, 849 Search PubMed; (c) I.-H. Park, R. Medishetty, S. S. Lee and J. J. Vittal, Chem. Commun., 2014, 50, 6585 RSC; (d) M. M. Unterlass, F. Emmerling, M. Antoniettia and J. Webera, Chem. Commun., 2014, 50, 430 RSC; (e) L. Dou, Y. Zheng, X. Shen, G. Wu, K. Fields, W.-C. Hsu, H. Zhou, Y. Yang and F. Wudl, Science, 2014, 343, 272 CrossRef CAS PubMed; (f) C. D. Papaspyrides and S. N. Vouyiouka, Solid State Polymerization, John Wiley & Sons, Inc., 2009 Search PubMed.
- (a) Y. Yin, Z. Li, J. Jin, C. Tusy and J. Xia, Synth. Met., 2013, 175, 97 CrossRef CAS PubMed; (b) M. Belletête, S. Beaupre, J. Bouchard, P. Blondin, M. Leclere and G. Durocher, J. Phys. Chem. B, 2000, 104, 9118 CrossRef.
- V. Lemau de Talance, M. Hissler, L.-Z. Zhang, T. Karpati, L. Nyulaszi, D. Caras-Quintero, P. Baeuerle and R. Reau, Chem. Commun., 2008, 2200 RSC.
- K. J. Hoffmann, L. Knudsen, E. J. Samuelsen and P. H. J. Carlsen, Synth. Met., 2000, 114, 161 CrossRef CAS.
- S. Ponomarenko, A. Muzafarov, O. Borshchev, E. Vodopyanov, N. Demchenko and V. Myakushev Russ, Chem. Bull., Int. Ed., 2005, 54, 684 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXTL, Version 6.14, Bruker Analytical X-ray Instruments, Inc, Madison, WI, USA, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- Y. Xia, A. G. MacDiarmid and A. J. Epstein, Macromolecules, 1994, 27, 7212 CrossRef CAS.
- D. Hohnholz, A. G. MacDiarmid, D. M. Sarno Jr and W. E. Jones, Chem. Commun., 2001, 2444 RSC.
- C. Kvarnström, H. Neugebauer, S. Blomquist, H. J. Ahonen, J. Kankare and A. Ivaska, Electrochim. Acta, 1999, 44, 2739 CrossRef.
- G. Louarn, J. Kruszka, S. Lefrant, M. Zagorska, I. Kulszewicz-Bayer and A. Pron, Synth. Met., 1993, 61, 233 CrossRef CAS.
- M. S. Agashe and C. I. Jose, J. Chem. Soc., Faraday Trans. 2, 1977, 1232 RSC.
- A. Paul, I. GiguEère and D. Liu, J. Chem. Phys., 1952, 20, 136 CrossRef PubMed.
- N. Zamoshchik and M. Bendikov, Adv. Funct. Mater., 2008, 18, 3377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, CIF files, crystallographic data, CV, TGA for corresponding polymers, 1H, 13C NMR and GPC profiles. See DOI: 10.1039/c4ra14915g |
| This journal is © The Royal Society of Chemistry 2015 |