
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical synthesis and characterization of novel samarium oxide nanoparticles: toward a heterogeneous Brønsted acid catalyst†
Gregory K.
Hodgson
a,
Stefania
Impellizzeri
a,
Geniece L.
Hallett-Tapley
ab and
Juan C.
Scaiano
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie, Ottawa, Ontario K1N 6N5, Canada. E-mail: Scaiano@photo.chem.uottawa.ca
bDepartment of Chemistry, St. Francis Xavier University, P.O. Box 5000, Antigonish, Nova Scotia B2G 2W5, Canada
First published on 2nd December 2014
Abstract
Samarium oxide nanoparticles (Sm2O3NP) were prepared photochemically for the first time. Characterization shows spherical, polydisperse Sm2O3NP stabilized by 4-HEBA, a substituted benzoic acid. The Sm2O3NP also possess Brønsted acidity. This new material may prove to be a potent heterogeneous acid catalyst.
In the ongoing pursuit of new and useful catalytic materials, nanochemistry has become a popular strategy for discovery and innovation. Widespread research has led to a library of nanoparticle (NP) synthesis techniques, and cutting-edge photochemical methods have recently provided environmentally benign, cost-effective synthetic routes.1,2 Many nanomaterials consist of well-characterized components possessing catalytic properties that can only be accessed at the nanoscale. In this context, gold and silver NP and nanoclusters are prime examples.3 Other nanocatalysts are modelled after well-performing bulk metal catalysts in an effort to further increase efficiency.4 The lanthanide series remains relatively unexplored, and represents a potentially untapped resource for the development of new nanostructures with as-yet undocumented catalytic properties. Samarium-based compounds may present such an opportunity. As an element, samarium is actually quite abundant5 and already has some niche applications (e.g. samarium–cobalt magnets).6 Samarium triflate is a potent Lewis acid catalyst,7 and SmI2 has been utilized extensively as a versatile reducing agent for single electron transfer reactions.8 Other samarium-based homogeneous catalysts have been employed in the degradation of polychlorinated biphenyls9 and in the dehydration of alcohols.10 Bulk samarium oxide catalyzes the oxidation of methane, ethane and ethylene.11–13 However, little is known about the behaviour of samarium and its oxides (Sm2O3 and SmO) at the nanoscale. Of the few examples of samarium oxide NP synthesis in the literature,13,14 lengthy procedures, safety concerns and supercritical conditions are obvious disadvantages. Faster, safer, environmentally friendly synthetic strategies are required if the potential to use these and other lanthanide-based materials for catalysis is to be investigated further. Here we report a simple photochemical route to novel samarium oxide nanoparticles (Sm2O3NP) possessing physicochemical properties that have the potential to make this new nanomaterial a potent heterogeneous Brønsted acid catalyst.
Novel Sm2O3NP were prepared photochemically, by UVA irradiation of the benzoin Irgacure-2959™ (I-2959) photoinitiator in the presence of samarium nitrate hexahydrate (Scheme 1). Similar mechanisms have been used to describe the photochemical synthesis of a variety of metallic and metal-oxide nanostructures.1 For example, cobalt oxide NP have been prepared by initial photoreduction of CoCl2 using Irgacure-907,15 followed by air oxidation of the cobalt nanoparticles. However, samarium oxidizes much more readily than cobalt and thus we believe that it is never fully reduced to Sm0. Although a millimolar concentration of photoinitiator would result in cessation of ketyl radical generation after only minutes of irradiation, a precipitate did not form until much later, at which point the partially reduced samarium precursor had been oxidized to Sm2O3NP. Dynamic Light Scattering (DLS) was used to monitor the NP growth over time, and indicated an initial stage of rapid growth followed by slower growth over the course of several hours (Fig. S1, ESI†). This experiment demonstrated that oxygen is required for the reaction, and also that reduction and oxidation occur concurrently during the initial phase of Sm2O3NP formation.
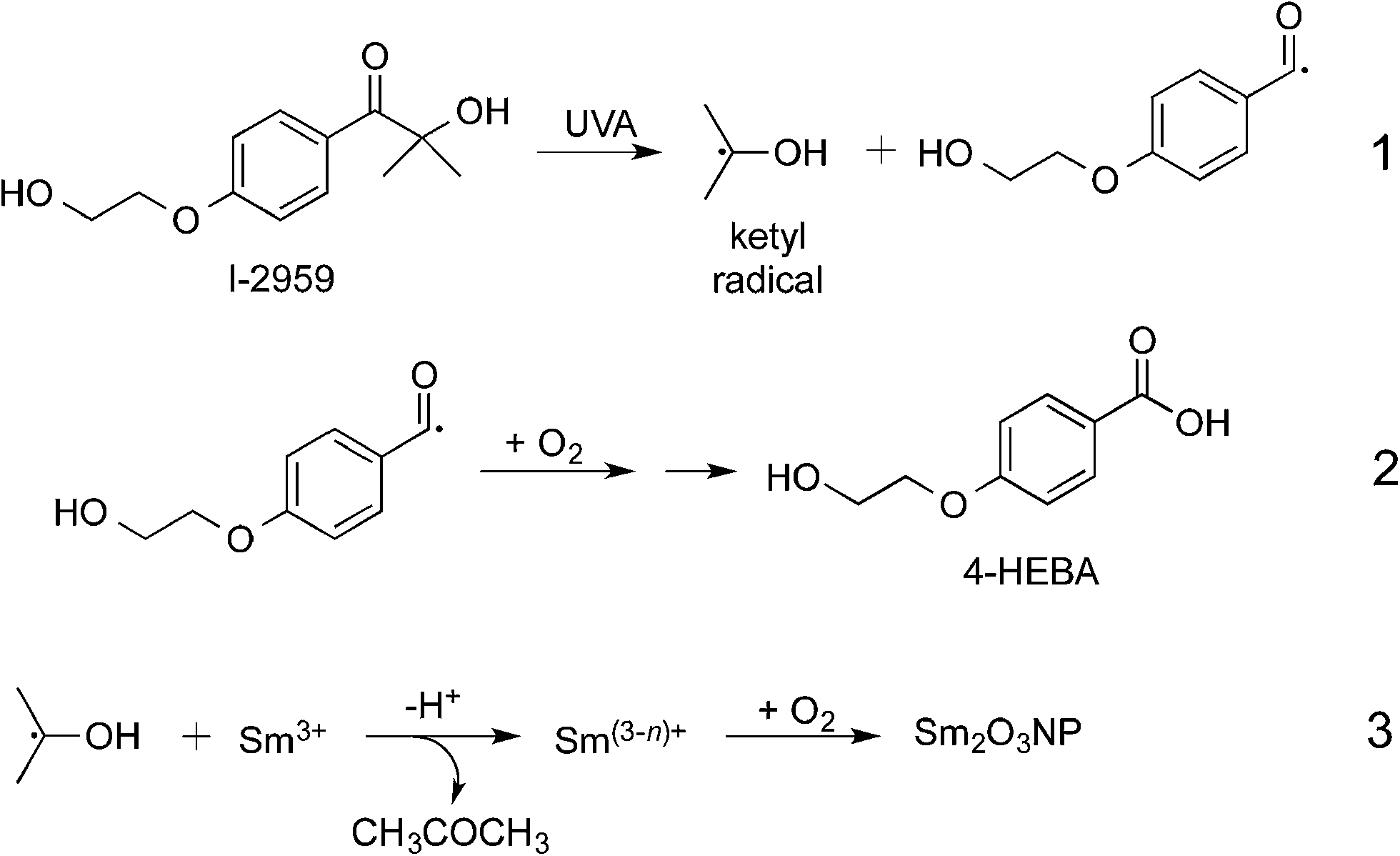 | ||
| Scheme 1 Photochemical preparation of Sm2O3NP in CH3CN. The small arrow in eqn (2) denotes the eventual reduction of the intermediate to 4-HEBA. In eqn (3), n equals 1 or 2 but not 3, as metallic samarium has not been observed. | ||
This procedure yielded a flaky brown-orange solid that rapidly settles out of many common solvents, but that is easily suspended in strong, polar aprotic solvents such as DMF and DMSO. For example, zeta potential measurements gave an average value of +23.1 mV in DMSO, indicating moderate colloidal stability. Energy Dispersive X-ray Spectroscopy (EDS) identified the primary constituents of the material to be samarium and oxygen (Fig. S2, ESI†).
X-ray Photoelectron Spectroscopy (XPS) detected samarium exclusively in the +3 oxidation state, confirming that the material is comprised of Sm2O3. This was evident from the presence of a doublet that dominated the 1050.0–1150.0 eV region of the XPS survey of the material (Fig. S3, ESI†). Peak splitting is well known to be the result of j–j coupling, which in this case gave rise to two intense peaks centred at 1084.0 and 1110.3 eV. These binding energies (BE) correspond to the 3d5/2 and 3d3/2 states of Sm3+ present in Sm2O3, respectively,13,16–20 consistent with the facile oxidation of samarium to Sm2O3 – the more stable of the two oxides.18–20 Further, no direct evidence of Sm2+ was obtained (a detailed interpretation of all XPS results is given in the ESI†). Traces of SmO could nonetheless be present, but it would exist as a transient surface species and represent only a minute fraction of the material's composition at any given time.11,13,17,21 Samarium is redox active, so it is possible that the material may respond to its chemical environment by alternating between Sm2O3 and SmO to some extent. In any event, quantification of the core level Sm 3d peak data revealed that the material contains roughly 40% samarium by mass.
X-ray diffraction (XRD) showed broad peaks roughly consistent with bulk phase Sm2O3 (Fig. S4, ESI†).10,13,22 Peak broadening is a direct result of NP formation and is commonly associated with amorphous solids.13,17,22,23 SEM revealed remarkably spherical, polydisperse particles with a mean diameter of 417 ± 114 nm (Fig. 1). This value was obtained by manually sizing 450 individual NP from a single SEM image using ImageJ software (Fig. S5 ESI†). Dynamic Light Scattering (DLS) performed on the same batch of Sm2O3NP (in DMSO) gave a larger mean diameter of 510 ± 122 nm (Table S1, ESI†). Although the magnitudes of the standard deviation in the SEM and DLS results put the two values within range of one another, the mean hydrodynamic diameter being greater than the mean diameter obtained by SEM analysis allows for the possibility that ligands may be coordinated to the NP surface. The most likely candidate for such a stabilizer is 4-(2-hydroxyethoxy)-benzoic acid (4-HEBA) formed during Sm2O3NP synthesis (Scheme 1). This compound has previously been identified as a photoproduct of I-2959 and is known to contribute to NP stability. The formation of 4-HEBA under ambient conditions is generally considered to involve trapping of the acyl radical by oxygen, formation of an intermediate peracid, and eventual reduction to 4-HEBA.11H NMR spectroscopy performed on Sm2O3 dissolved in DMSO-d6 detected 4-HEBA even after extensive washing (Fig. S6 and S7 ESI†). No other organic species were detected in the 1H NMR spectrum but elemental analysis concluded that the material is comprised of 38% carbon and 4.5% hydrogen (Table S2, ESI†). The presence of 4-HEBA accounts for the 38% carbon, which was also qualitatively detected by XPS. However, the molar quantity of 4-HEBA could not be reliably determined from core level C 1s XPS data due to probable sample contamination from adsorbed atmospheric carbon that could enhance the measured intensity of the C 1s peak.
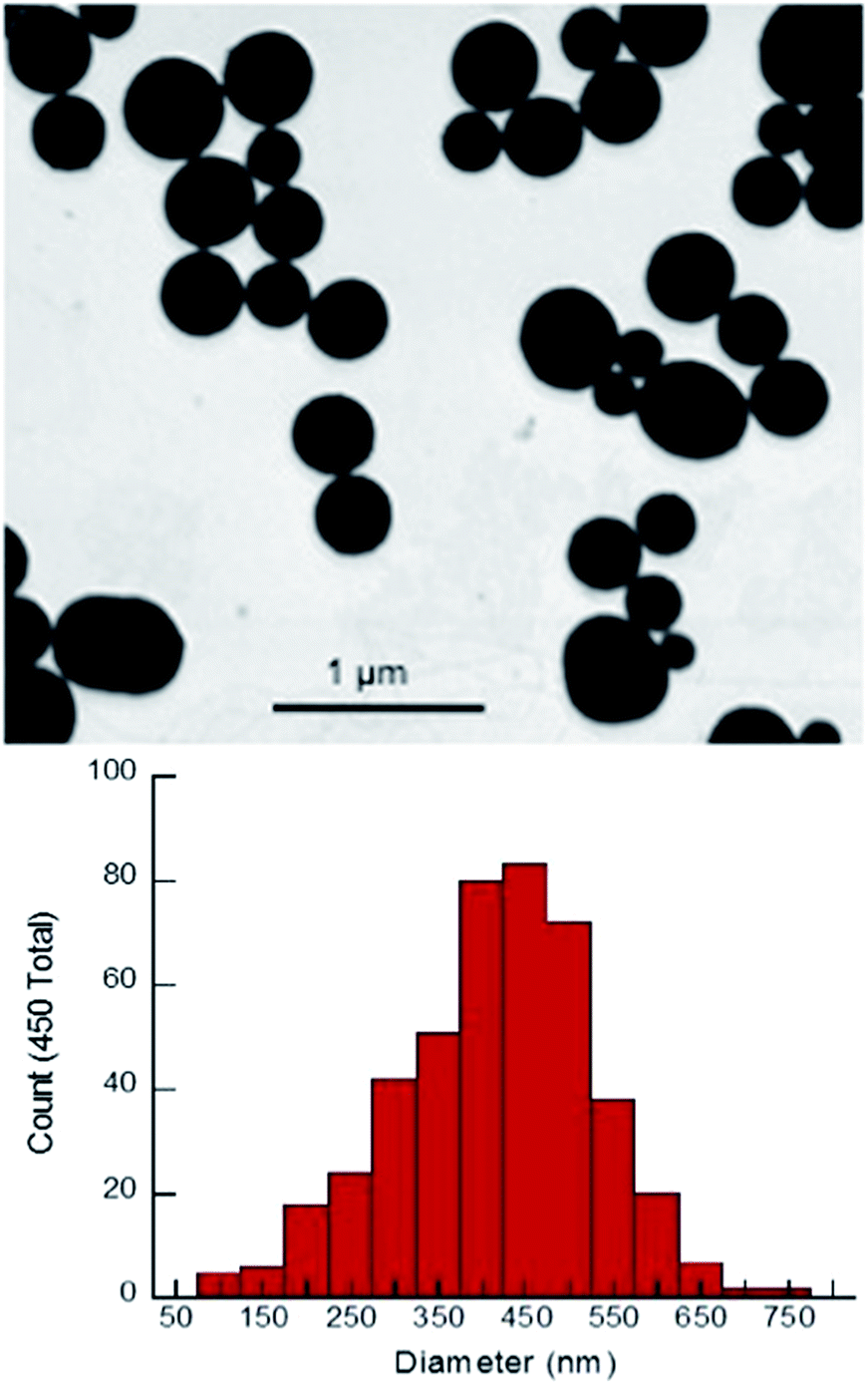 | ||
| Fig. 1 Upper panel: SEM image of Sm2O3NP. Lower panel: histogram showing the size distribution of Sm2O3NP based on manual analysis of SEM results. | ||
In order to ensure that the individual Sm2O3NP shown in Fig. 1 are not comprised of smaller NP subunits, TEM imaging was performed and showed no evidence of any internal structure or defects in the NP surface (Fig. S8, ESI†). Efforts to decrease the average size and polydispersity of the Sm2O3NP by altering the synthetic conditions were unsuccessful. Similarly, any NP too small to be obtained via centrifugation of the post-irradiation solution could not be harvested using a non-solvent approach; adding an excess of toluene to a concentrated volume of supernatant after centrifuging out the larger Sm2O3NP did not result in precipitation, even after several days at 4 °C. Laser drop ablation of a suspension of Sm2O3NP in MilliQ H2O did produce a small number of NP of diameter less than 50 nm but did not reduce the level of polydispersity (Fig. S9, ESI†). The optimal conditions for laser drop ablation, subsequent washing of the sample and the overall efficiency of the process require further investigation, and will be reported along with any observed effects of size and polydispersity upon the catalytic activity of Sm2O3NP.
Since 4-HEBA is only mildly acidic (pKa ≈ 4), its presence alone does not explain the level of acidity possessed by Sm2O3NP. Hammett indicator studies conducted using a 0.1% w/v solution of dicinnamalacetone (DCA) in toluene suggested that the Sm2O3NP have a pKa ≤ −3. Unfortunately, other common indicators with pKa values less than −3, such as benzalacetophenone (pKa −5.6) and anthraquinone (pKa −8.2), are colourless in the base form and yellow in the acid form.25 The colour change is undetectable when these indicators are exposed to the brown-orange Sm2O3NP. Therefore, the amount by which the pKa of Sm2O3NP falls below −3 cannot be experimentally determined using the Hammett indicator method. However, since DCA changes from yellow to red upon exposure to an acid, the total number of acid sites per gram of material can be estimated by titration of the solid acid with n-butylamine following exposure to the indicator. This experiment required 40 μL 0.1 M n-butylamine (in toluene) to titrate 5 mg of Sm2O3NP previously exposed to 1 mL 0.1% w/v DCA in toluene, corresponding to a total acid strength of 0.8 mmol g−1.
Since the titration method does not differentiate between Brønsted and Lewis acid sites, the acidity of the Sm2O3NP was also investigated using Fourier transform infrared (FTIR) spectroscopy. By comparing the FTIR spectrum of a solid acid before and after the adsorption of pyridine, the presence of Brønsted acid sites can be detected. With the correct experimental setup, the number of acid sites of each type can also be quantified by this method. Peaks in the FTIR spectrum in close proximity to 1540 cm−1 and 1440 cm−1 can often be attributed to the pyridinium ion formed upon adsorption of pyridine to Brønsted and Lewis acid sites, respectively.25 As shown in Fig. 2, saturation of Sm2O3NP with pyridine vapours resulted in the appearance of a band at 1540 cm−1, indicating the presence of Brønsted sites strong enough to interact with pyridine. The full-scale FTIR spectra of Sm2O3NP before and after exposure to pyridine are available in the ESI (Fig. S10 and S11†).
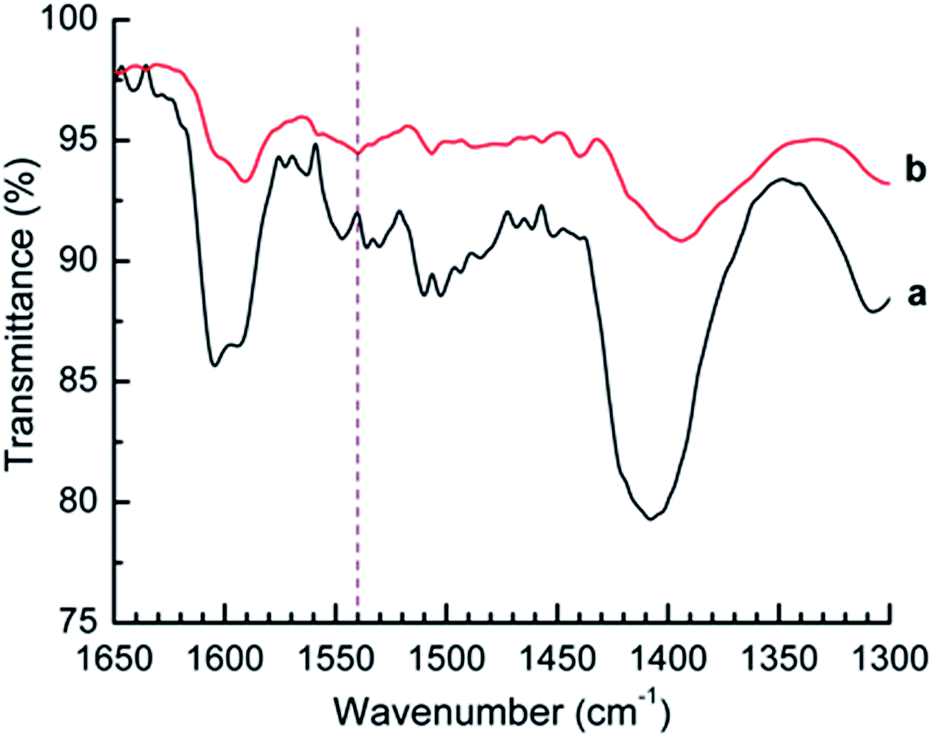 | ||
| Fig. 2 FTIR spectrum of Sm2O3NP before (a) and after (b) saturation with pyridine vapours. The vertical dashed line at 1540 cm−1 denotes the position of the characteristic pyridinium ion peak attributable to pyridine adsorbed onto Brønsted acid sites. | ||
In this case the small signal at 1440 cm−1 is not evidence of Lewis acid sites; the FTIR spectrum of pyridine showed a strong signal in that same position (Fig. S12, ESI†). However, the latter does not contain any signal in 1490–1570 cm−1 region, supporting evidence for the presence of Brønsted acid sites on the surfaces of Sm2O3NP. Overall, the Hammett acid indicator test, n-butylamine titration and pyridine adsorption experiments collectively demonstrated that the Sm2O3NP have a pKa ≤ −3, a total acid strength in the vicinity of 0.8 mmol g−1 and possess some degree of Brønsted acidity.
As a proof-of-concept, we show that Sm2O3NP can efficiently protonate the halochromic coumarin–oxazine molecular assembly 1. The absorption spectrum of 1 in CH3CN shows a band centred at 410 nm. The addition of an acid opens the oxazine ring and generates the stable fluorescent compound 2 (Fig. S13, ESI†). Within this transformation, the coumarin functionality is brought into conjugation with the cationic unit, bathochromically shifting the absorption band of the generated species by 180 nm.23,24 A fluorescence band centred at 645 nm can then be observed by selectively exciting 2 at λEx 570 nm. Therefore, the transformation of 1 into 2, promoted by the addition of a Brønsted acid, can be exploited in order to activate fluorescence and thus permits the investigation of materials with distinctive acidic properties using a simple experimental setup. In this case catalytic conversion to the ring-open form 2 began shortly after exposure to Sm2O3NP and was complete within 30 min (Fig. 3 and S14, ESI†). This confirmed that Sm2O3NP possess Brønsted acidity, a property that could make Sm2O3NP a useful heterogeneous acid catalyst.
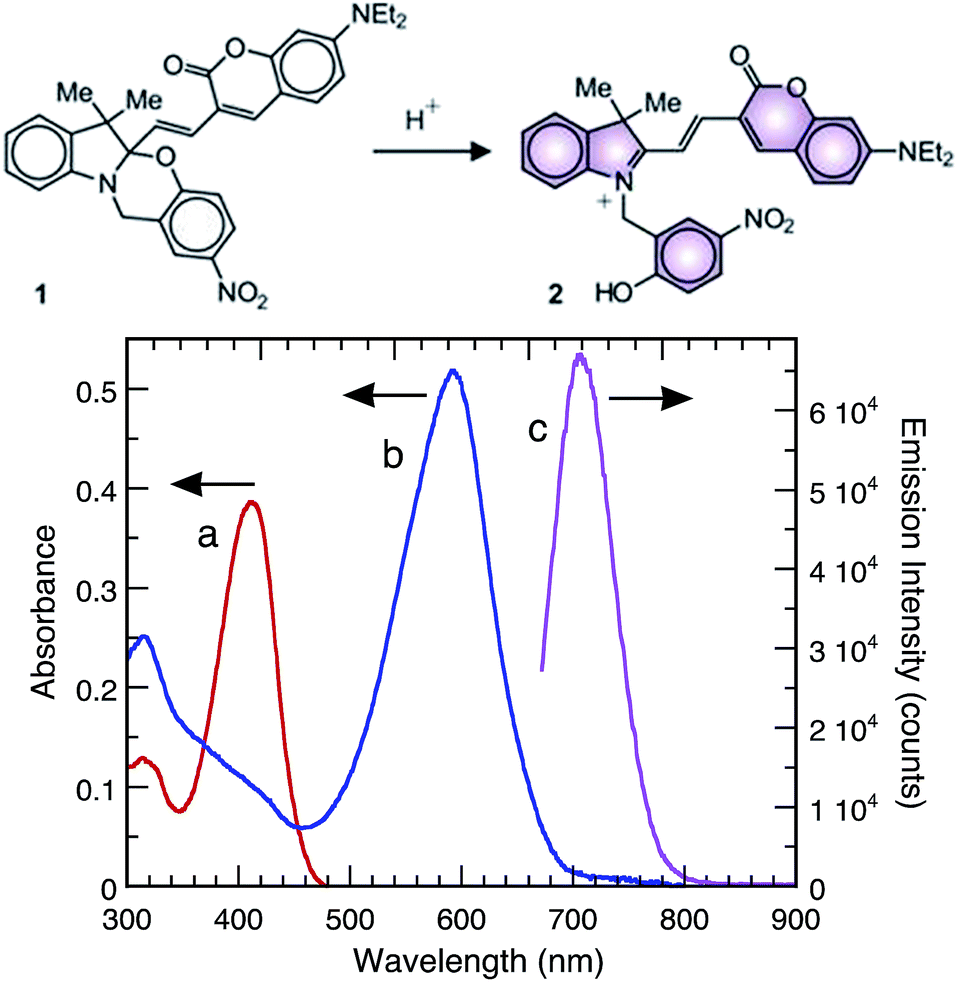 | ||
| Fig. 3 Upper panel: ring-opening of the halochromic switch. Lower panel: absorption spectra of 1 (10 μM, CH3CN, 25 °C) before (a) and after (b) 30 min exposure to Sm2O3 NP and subsequent centrifugation. Emission spectrum ((c) λEx = 570 nm, CH3CN, 25 °C) of 1 after 30 min exposure to Sm2O3 NP and subsequent centrifugation. | ||
In order to confirm that the observed Brønsted acidity of the Sm2O3NP is a surface effect, the impact of exposing Sm2O3NP to a strong base was evaluated. Sm2O3NP previously used to convert 1 to 2 were washed with CH3CN, treated with 2 mM NaOH three times, washed again with CH3CN and finally exposed to a new 10 μM solution of the closed-ring species 1. No conversion from 1 to 2 was observed, even after 24 h (Fig. S15, ESI†). However, the structural integrity of the Sm2O3NP was retained (Fig. 4). This would not be anticipated if protons were being leached from within the NP interior. Interestingly though, the surfaces of base-treated Sm2O3NP shown in Fig. 4 appear roughened or non-uniformly pitted. This may indicate the disruption of several surface oxide layers in close proximity to heterogeneously distributed Brønsted acid sites. In any case, these results confirm the surface acidity of the Sm2O3NP.
 | ||
| Fig. 4 SEM image of Sm2O3NP after repeated exposure to 2 mM NaOH and subsequent washing with CH3CN. | ||
Conclusions
We describe a photochemical approach to the synthesis of a novel lanthanide-based nanomaterial – Sm2O3NP – under very mild conditions. To the best of our knowledge, this is the first report of photochemically prepared Sm2O3NP in the literature. Not only are such methods beneficial from an environmental standpoint, improvements upon traditional synthetic strategies provided by photochemical techniques are necessary to achieve time- and cost-effectiveness that facilitates streamlined production of prototype materials. This in turn permits economic exploration of less than well-travelled regions in terms of the iterative design of new catalytic materials. A thorough characterization of the physicochemical properties of the Sm2O3NP reported here revealed spherical particles with a 4-HEBA ligand. More importantly, the Sm2O3NP possess surface Brønsted acidity, with a total acid strength of approximately 0.8 mmol g−1. This property endows the new material with potential as a Brønsted acid catalyst, as illustrated by the 1 → 2 conversion. We envision the eventual replacement of harsh homogeneous acid catalysts with Sm2O3NP and similarly designed heterogeneous nanocatalysts offering ease of separation and/or recyclability while maintaining high catalytic efficiency.Acknowledgements
The authors thank the Natural Sciences and Engineering Research Council, the Canada Foundation for Innovation and the Canada Research Chairs program. S. Impellizzeri acknowledges the award of a Banting Postdoctoral Fellowship.Notes and references
- K. L. McGilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef CAS PubMed; J. C. Scaiano, J. C. Netto-Ferreira, E. Alarcon, P. Billone, C. J. Bueno-Alejo, C. L. Crites, M. Decan, C. Fasciani, M. Gonzalez-Bejar, G. Hallett-Tapley, M. Grenier, K. L. McGilvray, N. L. Pacioni, A. Pardoe, L. Rene-Boisneuf, R. Schwartz-Narbonne, M. J. Silvero, K. Stamplecoskie and T. Wee, Pure Appl. Chem., 2011, 83, 913–930 CrossRef; J. C. Scaiano, K. G. Stamplecoskie and G. L. Hallett-Tapley, Chem. Commun., 2012, 48, 4798–4808 RSC; D. Malyshev, F. Bosca, C.-O. L. Crites, G. L. Hallett-Tapley, J. C. Netto-Ferreira, E. I. Alarcon and J. C. Scaiano, Dalton Trans., 2013, 42, 14049–14052 RSC; J. C. Scaiano, K. G. Stamplecoskie, K. L. McGilvray and N. L. Pacioni, A paradigm for the radical-mediated photochemical synthesis of metal nanostructures, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. Armido Studer and C. Chatgilialoglu, 2012, vol. 4, Polymers and Materials, ch. 73, pp. 2197–2210 CrossRef; E. Alarcon, C. J. Bueno-Alejo, C. W. Noel, K. G. Stamplecoskie, N. L. Pacioni, H. Poblete and J. C. Scaiano, J. Nanopart. Res., 2013, 15, 1374 CrossRef.
- C. J. Bueno-Alejo, C. D'Alfonso, N. L. Pacioni, M. González-Béjar, M. Grenier, O. Lanzalunga, E. I. Alarcon and J. C. Scaiano, Langmuir, 2012, 28, 8183–8189 CrossRef CAS PubMed.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed; K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef PubMed.
- G. A. Rance, W. A. Solomonsz and A. N. Khlobystov, Chem. Commun., 2013, 49, 1067–1069 RSC; F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Eur. J. Org. Chem., 2010, 2010, 1875–1884 CrossRef CAS; B. S. P. Anil Kumar, K. Harsha Vardhan Reddy, B. Madhav, K. Ramesh and Y. V. D. Nageswar, Tetrahedron Lett., 2012, 53, 4595–4599 CrossRef PubMed; R. Hudson, C.-J. Li and A. Moores, Green Chem., 2012, 14, 622–624 RSC; A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 Search PubMed; M. R. Decan, S. Impellizzeri, M. L. Marin and J. C. Scaiano, Nat. Commun., 2014, 5, 4612 Search PubMed.
- J. Gambogi, M. L. Jackson and L. D. Miller, U.S. Geological Survey Minerals Yearbook: Rare Earths, USGS, 2011 Search PubMed.
- C. M. Cooper, J. Wallace, M. Brookhart, M. Clark, C. Collins, W. X. Ding, K. Flanagan, I. Khalzov, Y. Li, J. Milhone, M. Nornberg, P. Nonn, D. Weisberg, D. G. Whyte, E. Zweibel and C. B. Forest, Phys. Plasmas, 2014, 21, 013535 CrossRef CAS PubMed; A. Haider, S. Akhtar, Z. Ahmad and M. Farooque, Key Eng. Mater., 2010, 442, 250–254 CrossRef.
- P. B. Gorepatil, Y. D. Mane and V. S. Ingle, Synlett, 2013, 24, 2241–2244 CrossRef CAS PubMed; A. V. Narsaiah, A. R. Reddy, B. V. S. Reddy and J. S. Yadav, Synth. Commun., 2010, 40, 1750–1757 CrossRef; A. V. Narsaiah, A. R. Reddy and J. S. Yadav, Synth. Commun., 2011, 41, 262–267 CrossRef.
- M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed.
- S. A. Jackman, C. J. Knowles and G. K. Robinson, Chemosphere, 1999, 38, 1889–1900 CrossRef CAS.
- G. A. M. Hussein, D. J. Buttrey, P. DeSanto Jr, A. A. Abd-Elgaber, H. Roshdy and A. Y. Z. Myhoub, Thermochim. Acta, 2003, 402, 27–36 CrossRef CAS.
- A. Ekstrom and J. A. Lapszewicz, J. Am. Chem. Soc., 1988, 110, 5226–5228 CrossRef CAS.
- R. V. Siriwardane, Langmuir, 1991, 7, 497–502 CrossRef CAS; V. T. Amorebieta and A. J. Colussi, J. Am. Chem. Soc., 1996, 118, 10236–10241 CrossRef.
- T.-D. Nguyen, D. Mrabet and T.-O. Do, J. Phys. Chem. C, 2008, 112, 15226–15235 CAS.
- J. Gao, Y. Zhao, W. Yang, J. Tian, F. Guan, Y. Ma, J. Hou, J. Kang and Y. Wang, Mater. Chem. Phys., 2003, 77, 65–69 CrossRef CAS; E. Reverchon, G. D. Porta, D. Sannino, L. Lisi, P. Ciambelli, B. Delmon, P. A. Jacobs, R. Maggi, J. A. Martens, P. Grange and G. Poncelet, Surf. Sci., 1998, 118, 349–358 Search PubMed; W. Zhu, L. Xu, J. Ma, R. Yang and Y. Chen, J. Colloid Interface Sci., 2009, 340, 119–125 CrossRef PubMed.
- T. Wee, B. D. Sherman, D. Gust, A. L. Moore, T. A. Moore, Y. Liu and J. C. Scaiano, J. Am. Chem. Soc., 2011, 133, 16742–16745 CrossRef CAS PubMed.
- M. G. Mason, S. T. Lee, G. Apai, R. F. Davis, D. A. Shirley, A. Franciosi and J. H. Weaver, Phys. Rev. Lett., 1981, 47, 730–733 CrossRef CAS.
- T.-D. Nguyen, C.-T. Dinh and T.-O. Do, Langmuir, 2009, 25, 11142–11148 CrossRef CAS PubMed.
- D. Cheng, Q. Xu, Y. Han, Y. Ye, H. Pan and J. Zhu, J. Chem. Phys., 2014, 140, 094706 CrossRef PubMed.
- M. Juel, B. T. Samuelsen, M. Kildemo and S. Raaen, Surf. Sci., 2006, 600, 1155–1159 CrossRef CAS PubMed; M. Kuchowicz and J. Kocaczkiewicz, Surf. Sci., 2008, 602, 3721–3727 CrossRef PubMed.
- Q. Xu, S. Hu, D. Cheng, X. Feng, Y. Han and J. Zhu, J. Chem. Phys., 2012, 136, 154705 CrossRef PubMed.
- D. D. Sarma, M. S. Hegde and C. N. R. Rao, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 1509–1520 RSC.
- L. Shi, G. Dong and D. He, Catal. Commun., 2007, 8, 359–365 CrossRef CAS PubMed.
- M. L. Marin, G. L. Hallett-Tapley, S. Impellizzeri, C. Fasciani, S. Simoncelli, J. C. Netto-Ferreira and J. C. Scaiano, Catal.: Sci. Technol., 2014, 4, 3044–3052 Search PubMed.
- E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 2010, 1, 3506–3509 CrossRef CAS; S. Swaminathan, M. Petriella, E. Deniz, J. Cusido, J. D. Baker, M. L. Bossi and F. M. Raymo, J. Phys. Chem. A, 2012, 116, 9928–9933 CrossRef PubMed.
- M. Yurdakoc, M. Ackay, Y. Tonbul and K. Yurdakoc, Turk. J. Chem., 1999, 23, 319–327 Search PubMed; H. A. Benesi, J. Phys. Chem., 1957, 61, 970–973 CrossRef CAS; D. T. Yazici and C. Bilgic, Surf. Interface Anal., 2010, 42, 959–962 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14841j |
| This journal is © The Royal Society of Chemistry 2015 |