DOI:
10.1039/C4RA14839H
(Communication)
RSC Adv., 2015,
5, 10134-10138
In situ synthesis of MoS2 on a polymer based gold electrode platform and its application in electrochemical biosensing
Received
19th November 2014
, Accepted 6th January 2015
First published on 6th January 2015
Abstract
Bulk layers of MoS2 were synthesized in situ on a polymer substrate at low temperature. The negative charges carried by the layered MoS2 are used to immobilize horseradish peroxidase conjugated IgG via the electrostatic attraction, forming an Au–MoS2/HRP hybrid. Trace H2O2 released from IgG-horseradish peroxidase was successfully evaluated in the linear range of 0–20 ng mL−1.
Introduction
Two-dimensional (2D) nanomaterials have attracted increasing attention because of their advantageous physical and chemical properties.1 In particular, 2D transition metal dichalcogenides (TMD) show a wide range of electronic, optical, mechanical, chemical, and thermal properties. In contrast to zero-band gap graphene, TMD molybdenum disulfide (MoS2) has an indirect band gap of 1.29 eV in the bulk state and it can be tuned from indirect to direct band gap (1.90 eV) through layer control. Moreover, in some cases MoS2 is utilized for the hydrodesulfurization reaction2 and for the hydrogen evolution reaction (HER) as a catalyst.1,3,4 Similarly, other 2D materials, such as graphene,5,6 epimeric monosaccharide-quinone hybrid7 on gold electrode have been utilized for electrochemical biosensors. Substantial efforts have been devoted to prepare thin layers of MoS2, by methods including adhesive tape-based micromechanical exfoliation,8 physical and chemical vapor deposition (CVD)9–11 and hydrothermal synthesis.12,13 Previous reports showed that an exfoliated single-layer MoS2 has a high mobility (200 cm2 V−1 s−1) with a hafnium oxide (HfO2) gate dielectric, and the on/off ratio of over 108 at room temperature,14,15 however MoS2 shows much low mobility (30–60 cm2 V−1 s−1) on Si/SiO2.16 It is difficult to achieve large grain size with high uniformity. While thermal chemical vapor deposition (CVD) method is the most extensive studied methods for synthesizing large and thin films of MoS2.10 Its applicability to flexible devices is limited because of the high (>600 °C) temperatures this method requires during the synthesis process for decomposition of carrier gases. Techniques that overcome this temperature issue to allow applications to flexible devices including growing MoS2 on a polymer substrate, which can be carried out at a process temperature below 300 °C or transfer of the MoS2 film from a Si/SiO2 substrate. The transfer step causes cracks or wrinkles in devices, so both methods have the potential to deteriorate MoS2 properties during the transfer process. Hence, there is a need to develop a new low temperature synthesis techniques that allow the direct synthesis of (in situ) MoS2 on the polymer substrate are needed to achieve high yield and performance.
Plasma enhanced chemical vapor deposition (PECVD) is well known for low temperature synthesis. During plasma synthesis, nanomaterials are subjected to various forces such as electrostatic forces, neutral drag, thermophoresis, ion drag in the sheath region, and photophoresis. These forces can be controlled by optimizing PECVD process parameters. In the past, our group succeeded in synthesizing mono-dispersed silicon nanoparticles with a uniform size and controlled morphology.17 Previous reports demonstrate synthesis of nanomaterials, such as carbon nanotubes,18 graphene,19 silicon nanoparticles and silicon nanowires,20 at temperatures below 500 °C using PECVD. The synthesis of nanomaterials at low temperatures is helped by the presence of radicals and dense sheaths (carrier gas and source gas) that facilitate the dissociation and decomposition of the gases. In this paper, we present an effective route for the in situ synthesis of MoS2 on a polymeric printed circuit boards (PCB). The in situ synthesis of MoS2 using PECVD is discussed in terms of the results of X-ray photoelectron spectroscopy (XPS), high resolution transmission electron microscopy (HR-TEM) and Raman scattering spectroscopy. Then the as-prepared MoS2 modified on Au electrode was used to immobilize IgG-HRP, and catalytic activity of the resultant HRP electrode to H2O2 reduction was observed with help of hydroquinone (HQ).
Experimental
Chemicals and materials
Phosphate buffer solution (pH = 7.0) was purchased from Sigma Aldrich (P4417, USA). IgG-HRP (NCl1430KR) was purchased from Thermoscientific. Hydroquinone (h3660) and hydrogen peroxide solution were purchased from Sigma Aldrich. The deionized water (DI water, >18 MΩ cm) used to prepare all solutions in this study was obtained from a water purification system. All chemicals were of reagent grade.
Fabrication of the Au electrode on PCB
We fabricated a layer of gold for all three working, counter, and reference electrodes to simplify fabrication process. Each gold (Au) electrode chip was fabricated by the semiconducting processes including positive photoresist (PR) coating, patterning, lift off and passivation in Scheme 1a. Approximately 250 three-electrode Au chips were fabricated on a glass wafer (4 inches in diameter). Au electrode chip with sensing PCB was fabricated by an electroplating method and used in further experiment. Each Au electrode chip was diced and glued on PCB.21 The center of Au electrode (diameter, 2 mm) was used further for the in situ synthesis of MoS2. The other two side Au electrodes were masked during synthesis. The centre Au electrode with MoS2 (Au–MoS2 electrode) was used as a working electrode during electrochemical cyclic voltammetry (CV) measurements.
 |
| | Scheme 1 Schematic of Au–MoS2 and IgG-HRP conjugated Au–MoS2 (a) Semiconductor processes for gold electrode fabrication (b) (1) 1 nm thick Mo is deposited on an Au electrode using an e-beam evaporator, (2) H2S + Ar plasma is generated and reacts with the Mo thin film in the PECVD chamber, (3) H2S+ combines or penetrates into the Mo thin film to form MoS2. (c) Schematic of the MoS2 biosensor device. For biosensing, the MoS2 is functionalized with receptors for specifically capturing target biomolecules. | |
In situ synthesis of layered MoS2 on the Au electrode PCB
Due to low carrier mobility, MoS2 itself is not suitable as an electrode material for device applications. There have been some studies on MoS2 FET based biosensor by transfer of MoS2 onto a metal electrode, which may be susceptible to contact resistance related issue. Thus, in the present research, was deposited by implementing PECVD in situ synthesis. MoS2 was synthesized using PECVD at 300 °C with H2S gas as a precursor on a prefabricated gold (Au) electrode on a glass wafer. The synthesis of MoS2 on a Si/SiO2 wafer under various process parameter conditions was reported previously.22 The process for the in situ synthesis of MoS2 is shown in Scheme 1b. The synthesis of MoS2 begins with the deposition of Mo metal on Au(111) herringbone surface by e-beam evaporation. The thickness of the molybdenum metal layer was 1 nm using a deposition rate below ∼0.1 Å s−1 under high vacuum conditions. During the deposition process, some of the Mo atoms were deposited on the surface of the Au electrode and some penetrated into the Au, forming an Au–Mo composite structure. This also facilitated a reduction of the contact resistance, which was opposite of that seen with the transfer of Mo on Au electrodes.23,24 During the PECVD process, the Au–Mo components reacted with the combination of H2S (source gas) and Ar (carrier gas) plasma in an ultra-high vacuum (UHV) at 300 °C. The synthesized MoS2 thin films were characterized by HR-TEM (JEOL-JEM ARM 200F), Raman spectroscopy (Alpha300 M+, WITec GmbH, Germany) wavelength and power of laser are 532 nm and 2 mW respectively and X-ray photoelectron spectroscopy (XPS, ESCA2000 (VG Microtech Inc.)).
Evaluation of H2O2 with IgG-HRP by cyclic voltammetry
For the H2O2 biosensor, HRP-conjugated IgG was immobilized on the Au electrode modified with MoS2 to form an Au–MoS2/HRP electrode via the drop casting method (Scheme 1c). The Au–MoS2 electrodes were washed with deionized water to eliminate unbound IgGs. The concentration of IgG-HRP immobilized on each Au–MoS2 electrode in these experiments were 0, 0.1, 0.5, 1, 5, 10 or 20 ng mL−1. Electrochemical immunoassay measurements were performed on a VersaSTAT 3 potentiostat/galvanostat (Ametek, USA) with the three electrodes comprising a platinum wire as the auxiliary electrode, Ag/AgCl (sat'd KCl) as the reference and a modified Au–MoS2 as the working electrode. The Au–MoS2/HRP electrodes were placed in an electrochemical cell containing 5.0 mL pH 7.0 PBS buffer, 0.5 mM hydroquinone (HQ) and 5.0 mM H2O2 where hydroquinone worked as a diffusional mediator. Eqn (1)–(4) shows the overall electron relay mechanism for hydrogen peroxide reduction via the immobilized HRP enzyme.25| | | H2O2 + HRP reduction → H2O + HRP oxidation | (1) |
| | | HRP oxidation + HQ reduction → HRP reduction + HQ oxidation | (2) |
| | | HQ oxidation + 2H + 2e− → HQ reduction | (3) |
| | | H2O2 + 2e− + 2H+ → 2H2O | (4) |
In the presence of HRP and hydroquinone (HQ), the addition of H2O2 triggered the oxidation of HQ to benzoquinone (BQ) which is subsequently reduced giving a current response of the electrode.
Results and discussion
Preparation and characterization of in situ Au–MoS2
In the plasma phase, which is an ionized gas phase, the H2S source gas is easily dissociated at low temperature. By taking advantage of this property, we synthesized wafer-scale layered MoS2 at 300 °C on the Au electrode deposited PCB, which is a polymeric substrate.
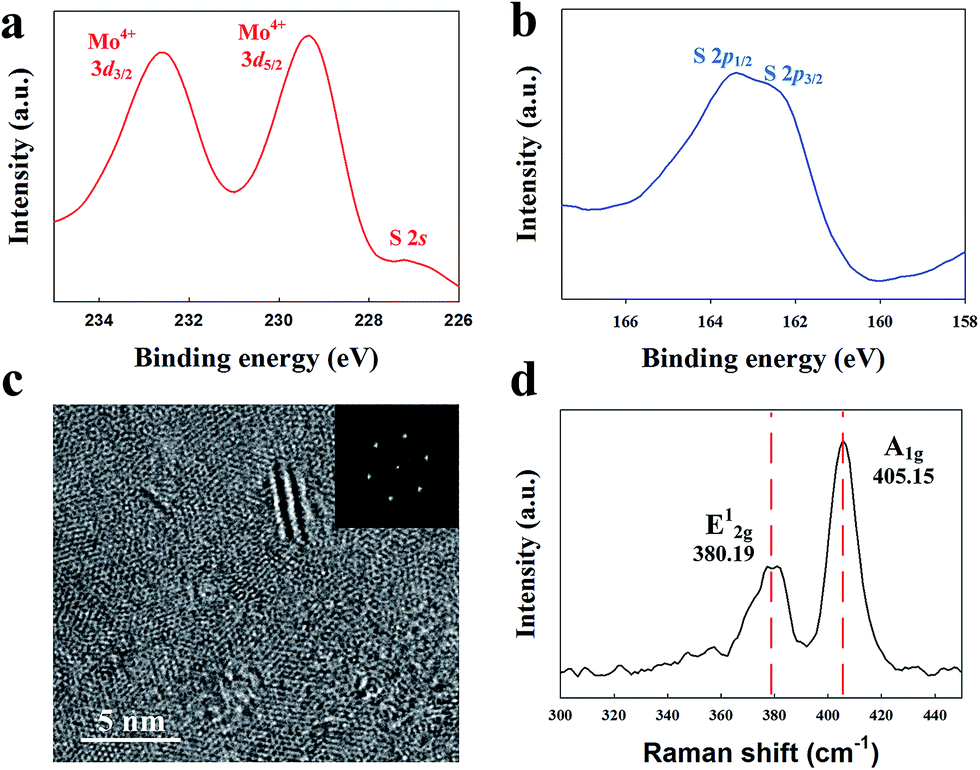
XPS was used to determine the chemical bonding composition of the surface. Fig. 1a and b show the XPS spectra of the MoS2 nanomaterial on Au electrodes. The Mo 3d spectrum (Fig. 1a) exhibited two well-separated peaks at binding energy levels of 229.3 eV and 232.6 eV respectively. The S 2p spectrum (Fig. 1b) shows two merged peaks at 162.5 eV and 163.4 eV. The XPS scans for other synthesis temperature conditions confirmed the chemical bonding states of the MoS2 layers. The calculated atomic concentration of Mo and S from XPS was 34.4% and 65.6%, yielding a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.90. The ratio from our PECVD-produced MoS2 is the same as the ratio of the CVD-synthesized MoS2.26
:1.90. The ratio from our PECVD-produced MoS2 is the same as the ratio of the CVD-synthesized MoS2.26
 |
| | Fig. 1 Chemical composition analysis of as-prepared MoS2 thin films using PECVD by X-ray photoemission spectroscopy (XPS) (a) Mo 3d and (b) S 2p region. (c) A typical HR-TEM image taken from the center of MoS2 at 300 °C. The insets are the selected area electron diffraction (SAED) (d) the Raman spectra, which is the E12g and A1g vibrational modes of MoS2 on an Au electrode. | |
Fig. 1c shows the morphology of the MoS2 thin film as obtained by analysing HR-TEM, transferred to the TEM grid (TED PELLA. INC, 200 nm silicon nitride membrane). The measured grain size at 300 °C ranged from 5–7 nm and was of a crystalline nature. Raman spectra (Fig. 1d) show that the MoS2 synthesized at 300 °C had E12g and A1g modes with observed Raman peaks at 380.15 and 405.15 cm−1. The 15 cm−1 difference between these peaks indicates the presence of about 6 to 7 layers of MoS2.27
Electrochemical investigation
The effect of scan rate on the anodic and cathodic peak currents at the Au–MoS2 electrode/HRP was investigated in 10 mM PBS (pH 7.0) using cyclic voltammetry (CV). As shown in Fig. 2a, it depicts the CV response of the Au–MoS2 electrode with different scan rate. The current value was observed in the absence of H2O2 which resulted from surface defects at 10 mV s−1 (curve 1). However, after the addition of 5 mM H2O2 at 10 mV s−1, the center of the reduction peak slightly increased to −0.25 V (curve 2). The redox peaks were clearly observed with a scan rate ranging from 10 to 100 mV s−1 demonstrating that the reduction and oxidation peak currents Increased as the scan rate increased. When the scan rate was changed 10 to 100 mV s−1, the reduction peak was proportional to the voltage from −0.25 to −0.29 V (curves 2, 3, and 4). The shift of the potential and the increase of the current indicated a notable enhancement of the electrocatalytic activity of the Au–MoS2 electrode toward the reduction of H2O2. Fig. 2b depicts the cyclic voltammetry (CV) response of the Au–MoS2 electrode conjugated with IgG-HRP with a scan rate of 100 mV s−1. Fig. 2b, curve 1, shows the CV response of the Au–MoS2 electrode in 0.1 M PBS, while curve 2 is response with the addition of 5 mM H2O2, and curve 3 shows the response of Au–MoS2/HRP (1 ng mL−1) in the presence of H2O2. The H2O2 reduction was promoted by the electrocatalysis from IgG-HRP immobilized on the Au–MoS2 electrode.
 |
| | Fig. 2 Cyclic voltammetry (CV) of MoS2 thin film deposited on Au electrode at different scan rate (a) (1) only 10 mM PBS without 5 mM H2O2 (used as buffer) at a scan rate of 10 mV s−1, (2) 5 mM H2O2 at a scan rate of 10 mV s−1, (3) 50 mV s−1, and (4) 100 mV s−1. The inset shows fabricated gold electrode chip on glass wafer and it mounted on printed circuit board (PCB). (b) (1) Only 10 mM PBS without H2O2 at a scan rate of 10 mV s−1, (2) 5 mM H2O2 at 100 mV s−1, (3) redox cycling that combines 1 ng mL−1 IgG-HRP at 100 mV s−1. | |
As shown in Fig. 3, well-characterized redox peak currents were observed at an IgG-HRP concentration ranging from 0 to 20 ng mL−1 with a scan rate of 100 mV s−1. The reduction current increased noticeably with the increase of IgG-HRP concentration. The inset indicated the linearity of the current level measured against the concentration of IgG-HRP from 0 to 20 ng mL−1 at a corresponding potential of −0.8 V. Moreover it was found that reduction peak currents were proportional to the concentrations of HRP, indicating typical electrochemical behaviour of immunoassays as well as a much larger peak current, which was observed because of the direct electron transfer between the HRP and underlying electrode, which was enhanced by the MoS2 thin film. In comparison with previous reported work the presented H2O2 sensor has lower detection limit28 and linear response.29 From these observations, the sensing mechanism of this sensor is the conjugation of the IgG target molecules and antibodies to IgG immobilized on Au–MoS2/immuno-substances by adsorption of HRP-conjugated IgG. By utilizing well-known sandwich based immunoassay, the detection efficiency of the targeted sample can be enhanced by the competitive reaction. Furthermore, the current level caused by antibody-target-detection can be increased by reducing the possibility of non-specific binding of the antibodies after pre-treatment of the bovine serum albumin (BSA) blocking step, this setup is able to detect nothing but targeted conjugation.
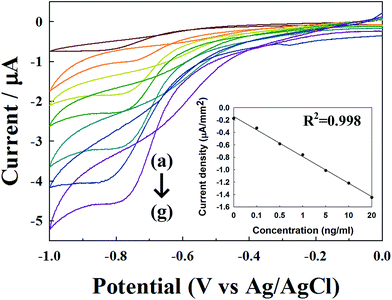 |
| | Fig. 3 CV of surface modification by MoS2 combine various concentration of IgG-HRP, (a) 0, (b) 0.1, (c) 0.5, (d) 1, (e) 5, (f) 10 and (g) 20 ng mL−1 at 100 mV s−1. The inset is the calibration curve of the current density to the concentration of IgG-HRP from 0 to 20 ng mL−1 at −0.8 V. | |
Conclusions
In summary, an in situ synthesis method for bulk layer of MoS2 was developed to fabricate H2O2 sensors. MoS2 was successfully synthesized at 300 °C on a polymeric PCB substrate. The resultant Au–MoS2/IgG-HRP electrode shows good electrocatalytic performance toward the reduction of H2O2 when HQ is used as mediator. The cyclic voltammetry results showed that the sensor of Au–MoS2 conjugated with IgG-HRP thus exhibited excellent analytical responses to H2O2 with a wide linear range. In particular, the linear range of IgG-HRP was measured to be 0–20 ng mL−1 with an R2 value of 0.998. The aim of this study to immobilize on Au–MoS2/immuno-substances, which can be used as an application in electrochemical biosensor. Our findings show that in situ synthesis process of MoS2 can be used as biocompatible matrix for the enzyme immobilization and construction of electrochemical biosensor. Moreover it can be expected to have the potential for commercial applications because of its low temperature in situ synthesis process and the ease at which other chemical and biosensors could be developed using this sensing platform.
Acknowledgements
This study was supported by the research fund of the Ministry of Trade, Industry, and Energy of Korea (grant no. 10045220). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2009-0083540).
Notes and references
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- E. Benavente, M. Santa Ana, F. Mendizábal and G. González, Coord. Chem. Rev., 2002, 224, 87–109 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Z. Li, S.-S. Deng, Y. Zang, Z. Gu, X.-P. He, G.-R. Chen, K. Chen, T. D. James, J. Li and Y.-T. Long, Sci. Rep., 2013, 3, 2293 Search PubMed.
- B.-W. Zhu, L. Cai, X.-P. He, G.-R. Chen and Y.-T. Long, Chem. Cent. J., 2014, 8, 1–6 CrossRef PubMed.
- X.-P. He, X.-W. Wang, X.-P. Jin, H. Zhou, X.-X. Shi, G.-R. Chen and Y.-T. Long, J. Am. Chem. Soc., 2011, 133, 3649–3657 CrossRef CAS PubMed.
- K. Novoselov, D. Jiang, F. Schedin, T. Booth, V. Khotkevich, S. Morozov and A. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
-
A. R. Lansdown, Molybdenum disulphide lubrication, Elsevier, 1999 Search PubMed.
- Y. H. Lee, X. Q. Zhang, W. Zhang, M. T. Chang, C. T. Lin, K. D. Chang, Y. C. Yu, J. T. W. Wang, C. S. Chang and L. J. Li, Adv. Mater., 2012, 24, 2320–2325 CrossRef CAS PubMed.
- Y. Zhan, Z. Liu, S. Najmaei, P. M. Ajayan and J. Lou, Small, 2012, 8, 966–971 CrossRef CAS PubMed.
- W.-J. Li, E.-W. Shi, J.-M. Ko, Z.-z. Chen, H. Ogino and T. Fukuda, J. Cryst. Growth, 2003, 250, 418–422 CrossRef CAS.
- Y. Peng, Z. Meng, C. Zhong, J. Lu, W. Yu, Y. Jia and Y. Qian, Chem. Lett., 2001, 772–773 CrossRef CAS.
- Z. Yin, H. Li, H. Li, L. Jiang, Y. Shi, Y. Sun, G. Lu, Q. Zhang, X. Chen and H. Zhang, ACS Nano, 2011, 6, 74–80 CrossRef PubMed.
- Y. Yoon, K. Ganapathi and S. Salahuddin, Nano Lett., 2011, 11, 3768–3773 CrossRef CAS PubMed.
- W. Bao, X. Cai, D. Kim, K. Sridhara and M. S. Fuhrer, Appl. Phys. Lett., 2013, 102, 042104 CrossRef PubMed.
- C. Ahn, K. Kim, H. Choi, A. Kulkarni and T. Kim, Thin Solid Films, 2011, 519, 7086–7089 CrossRef CAS PubMed.
- M. Meyyappan, L. Delzeit, A. Cassell and D. Hash, Plasma Sources Sci. Technol., 2003, 12, 205 CrossRef CAS.
- T.-O. Terasawa and K. Saiki, Carbon, 2012, 50, 869–874 CrossRef CAS PubMed.
-
M. Adachi and K. Karim, Photovoltaic Specialists Conference (PVSC), 2010 35th IEEE, 2010 Search PubMed.
- H. Y. Kim, K. J. Jang, M. Veerapandian, H. C. Kim, Y. T. Seo, K. N. Lee and M.-H. Lee, Biotechnol. Rep., 2014, 3, 49–53 CrossRef PubMed.
- H. Kim, C. Ahn, G. Arabale, C. Lee and T. Kim, ECS Trans., 2013, 58, 47–50 CrossRef PubMed.
- J. Lauritsen, S. Helveg, E. Lægsgaard, I. Stensgaard, B. Clausen, H. Topsøe and F. Besenbacher, J. Catal., 2001, 197, 1–5 CrossRef CAS.
- Z. Song, T. Cai, J. A. Rodriguez, J. Hrbek, A. S. Chan and C. M. Friend, J. Phys. Chem. B, 2003, 107, 1036–1043 CrossRef CAS.
- C. Camacho, J. C. Matías, B. Chico, R. Cao, L. Gómez, B. K. Simpson and R. Villalonga, Electroanalysis, 2007, 19, 2538–2542 CrossRef CAS PubMed.
- Y. Shi, J.-K. Huang, L. Jin, Y.-T. Hsu, S. F. Yu, L.-J. Li and H. Y. Yang, Sci. Rep., 2013, 3, 1839 Search PubMed.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS PubMed.
- S. HyunáMoh, RSC Adv., 2013, 3, 22940–22943 RSC.
- D. Liu, L. Yang, J. S. Huang, Q. H. Guo and T. Y. You, RSC Adv., 2014, 4, 13733–13737 RSC.
Footnote |
| † The authors pay equal contributions to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a