
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical spectroscopic studies on the complexation of stilbazolium dyes with a water soluble pillar[5]arene†
Márton
Bojtár
a,
Zoltán
Szakács
b,
Dóra
Hessz
c,
Miklós
Kubinyi
bc and
István
Bitter
*a
aDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, 1521 Budapest, Hungary. E-mail: bitter@oct.bme.hu
bDepartment of Physical Chemistry and Materials Science, Budapest University of Technology and Economics, 1521 Budapest, Hungary
cInstitute of Materials and Environmental Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, 1519 Budapest, Hungary
First published on 10th March 2015
Abstract
The host–guest interactions of a water soluble carboxylato-pillar[5]arene (WPA5) and three stilbazolium dyes (containing 9-anthryl, 1-pyrenyl and 4-dimethylaminophenyl groups) were studied by UV-Vis and fluorescence spectroscopy giving a detailed description of the spectral changes. A different spectroscopic response was observed in each case, with the most significant result of 28-fold fluorescence enhancement and intense color change in the case of 4-dimethylaminostyryl-N-methylpyridinium iodide (DAST, G3). In addition, a FID (fluorescence indicator displacement) system comprised of WPA5 and G3 was shown to detect paraquat by turn-off fluorescence in aqueous solution.
Introduction
Since the foundation of supramolecular chemistry1 the four generations of host molecules (cyclodextrins, crown ethers, calixarenes and cucurbiturils) have been the main targets in host–guest chemical studies. However, the recent discovery of pillararenes (PAs)2 has attracted great attention as this novel type of host molecules possess advantageous chemical (easy synthesis and modifications), structural (different cavity size, symmetry) and binding characteristics. Over the past three years numerous review papers3 discussing mainly the synthesis, conformations, chemical modifications and properties of pillararenes have been published. For years PA5 was the only representative of the family with acceptable synthetic availability.4 Lately a similarly simple access to PA6 was reported5 along with the identification and separation of some higher (up to 15) membered PAs.6 Although the parent PAs are sufficiently soluble in organic solvents to study their binding properties by spectroscopic methods, a break-through was achieved with water soluble pillararenes (WPAs)7,8 making studies possible in aqueous systems.A lot of pillararene-based host–guest interactions have been explored, especially with PA5,6 and WPA5,6. The guest molecules consist of mono- and diamines,9 ammonium, imidazolium, pyridinium salts,10N-methylacridinium iodide11 and basic amino acids.12 Among guest molecules, bipyridinium salts (paraquat and homologues) have been the most extensively studied and the complexations were primarily monitored by NMR, as generally neither PAs nor the guests were sensitively detectable with optical spectroscopic methods. A pillar[5]arene host with pyrene fluorescent marker,9 a quaternary ammonium attached to tetraphenylethene13 and an N-methylacridinium fluorescent guest are to be mentioned as exceptions.11 Beside the rare utilization of fluorescence detection in the complexation studies of PAs, the striking lack of stilbazolium dyes (mainly 4-styryl-N-methylpyridinium) among the numerous pyridinium guest molecules tested is surprising, albeit they are of great scientific and technologic interest as nonlinear optical materials,14 organelle-targeted fluorescent probes,15 norepinephrine transporter ligands,16etc.
In this paper the fluorescence spectroscopic investigations of host–guest interaction between WPA5 and stilbazolium dyes G1–G3 (see Fig. 1) in aqueous solution are presented. In addition, a fluorescence indicator displacement (FID) system comprised of WPA5 and DAST (4-dimethylaminostyryl-N-methylpyridinium iodide, G3) is shown to sensitively detect paraquat herbicide.
 | ||
| Fig. 1 Structures of WPA5 and guests G1–G3. | ||
Results and discussion
Stilbazonium dyes (G1–G3) were prepared by the piperidine-catalyzed condensation of the respective aldehydes with 1,4-dimethylpyridinium iodide according to previously reported methods.17WPA5 was synthesized as described recently.18 The complexation studies were performed primarily by UV-Vis and fluorescence spectroscopy in aqueous media.Complexation studies
Upon addition of WPA5 to the aqueous solution of G1, an increase in fluorescence intensity (Fig. S1†) was observed accompanied by a bathochromic shift of 18 nm. The maximum emission (F/F0 = 6.5 at 616 nm) was reached at 5 equivalent of WPA5, as it can be seen in Fig. S1.† The measurements were found, however, poorly reproducible presumably due to the E/Z-isomerism of G1 upon irradiation with the UV light.19 In this way, therefore, the complexation could not be evaluated quantitatively. Interestingly, an opposite emission behavior was experienced with G2, where ethanol–water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture was used for solubility reasons. Upon addition of WPA5 resulted in a slight decrease of fluorescence (Fig. S1†) which may be attributed to different emission mechanisms originated from the planarity of G2vs. the non-planar structure of G1.19 A Job's plot of the fluorescence intensities at 595 nm (see Fig. S2†) indicated the formation of an 1:1 complex, for which an association constant of K1 = (2.64 ± 0.2) × 105 M−1 was obtained by a least-square fitting of the fluorescence spectra (for details of calculations see Section 4 in ESI†). As G1 and G2 contain bulky aromatic fluorophore groups, we supposed the complex formation was simply driven by multiple electrostatic interactions between the anionic host and the cationic pyridinium moiety. NMR-studies carried out with G1 in D2O–DMSO-d6 = 1:1 mixture (for G2 we could not find a proper solvent mixture) revealed the proton signals in the complex were mostly unchanged with the exception of the signals of the pyridinium moiety (Fig. S5†). We observed large upfield shifts (approx. 1 ppm) and broadening of the signals of the N+Me and Py+-2,6-H and a slight upfield shift (0.3 ppm) assigned to the Py+-3,5-H protons. The results clearly show that the bulky 9-anthryl group prevent the complete inclusion of G1 and this assumption can be valid for the pyrene-substituted G2 as well.
:1 mixture was used for solubility reasons. Upon addition of WPA5 resulted in a slight decrease of fluorescence (Fig. S1†) which may be attributed to different emission mechanisms originated from the planarity of G2vs. the non-planar structure of G1.19 A Job's plot of the fluorescence intensities at 595 nm (see Fig. S2†) indicated the formation of an 1:1 complex, for which an association constant of K1 = (2.64 ± 0.2) × 105 M−1 was obtained by a least-square fitting of the fluorescence spectra (for details of calculations see Section 4 in ESI†). As G1 and G2 contain bulky aromatic fluorophore groups, we supposed the complex formation was simply driven by multiple electrostatic interactions between the anionic host and the cationic pyridinium moiety. NMR-studies carried out with G1 in D2O–DMSO-d6 = 1:1 mixture (for G2 we could not find a proper solvent mixture) revealed the proton signals in the complex were mostly unchanged with the exception of the signals of the pyridinium moiety (Fig. S5†). We observed large upfield shifts (approx. 1 ppm) and broadening of the signals of the N+Me and Py+-2,6-H and a slight upfield shift (0.3 ppm) assigned to the Py+-3,5-H protons. The results clearly show that the bulky 9-anthryl group prevent the complete inclusion of G1 and this assumption can be valid for the pyrene-substituted G2 as well.
When WPA5 was added to the aqueous solution of G3, intense optical responses were observed. Both the colorimetric (from yellow to orange) and fluorescent changes were visible to the naked eye (Fig. 2). In contrast, a significant fluorescence enhancement (F/F0 = 28 at 613 nm accompanied by a 20-fold increase in the fluorescence quantum yield) occurred even at low WPA5 concentrations (see Table 1 for spectroscopic details). The association constant for 1:1 comlex (see Fig. S3† for Job's plot) calculated from the absorption spectra was K1 = (1.3 ± 0.1) × 106 M−1. We speculated that the complex formation was reached by the penetration of G3 into the cavity of the host molecule driven by electrostatic attraction, hydrophobic interaction, π–π stacking and C–H⋯π interactions. The strong binding affinity in this system is attributed to the accumulation of these non-covalent forces. The inclusion phenomena were confirmed by 1H-NMR studies (Fig. 3). Because of the insufficient solubility of G3 in water, D2O–DMSO-d6 = 1:1 mixture was used as NMR solvent. Almost all signals deviated in the complex (1 equiv. G3). The most significant difference was observed in the case of the aromatic protons including the aminophenyl moiety as well: all the protons shifted upfield and/or broadened/disappeared from the spectrum which is attributed to the strong shielding by the electron-rich aromatic system surrounding the electron-deficient G3. Only the signals of the protons on the dimethylamino group did not show detectable changes. These changes were accompanied by the slight downfield shifts of the aromatic protons on the host WPA5 as well as the splitting of the O–CH2 protons into two sets of peaks indicating the restriction of swinging of the constituent units observed previously with paraquat.7 To characterize the effect of the DMSO co-solvent on the complex formation, the association constant K1 was also determined in water–DMSO 1:1 mixture from the absorption spectra. Its value, (1.5 ± 0.1) × 105 M−1, was lower than in neat water, showing the importance of the hydrophobic nature of the dye in the complex formation.
 | ||
| Fig. 2 Absorption and fluorescence spectra of G3 (5.0 μM) upon addition of WPA5 (0 to 1.46 equiv.) in water (excitation at 506 nm); inset: photographs showing colorimetric and fluorescent changes in aqueous solution (10.0 μM G3 (left vial), 10.0 μM G3 and 30.0 μM WPA5 (right vial)). Note: aqueous solution ofWPA5is colorless and therefore omitted. | ||
| λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ | τ F (ps) with rel. weights | |
|---|---|---|---|---|---|
| a Stimulated emission decay time from ref. 20, λpump = 400 nm, λprobe = 620 nm. b Calculated from the spectra of samples with constant G3 and varying WPA5 concentrations. c λ exc = 470 nm, λem = 610 nm. | |||||
| G3 | 449 | 29200 |
615 | 0.002 | 12 (100%)a |
| WPA5·G3 | 515b | 14300b |
607 | 0.041 | 310 (48%)c 640 (52%) |
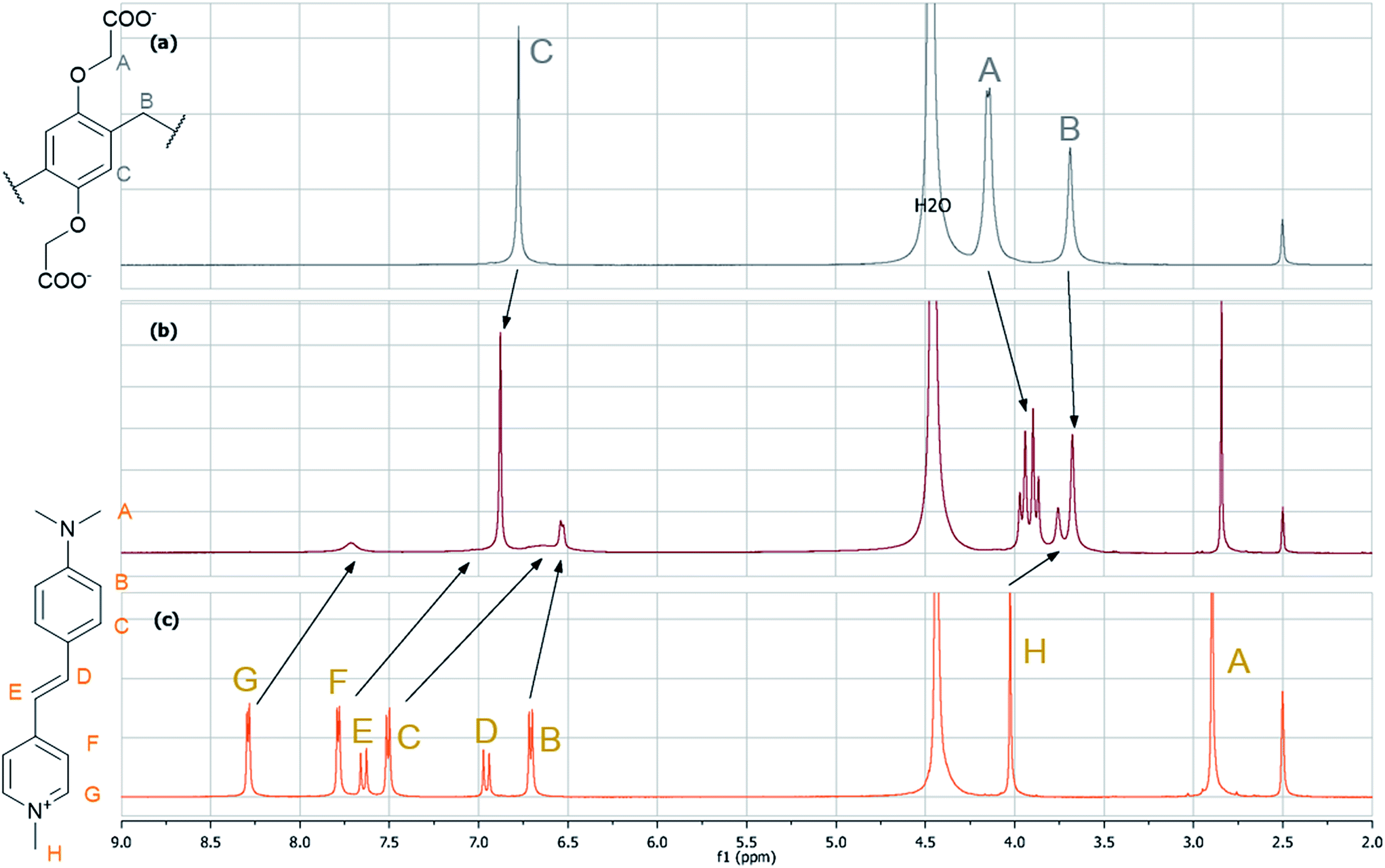 | ||
| Fig. 3 Partial 1H-NMR spectra (500 MHz, D2O–DMSO-d6 1:1, 298 K) of (a) WPA5; (b) G3 and WPA5 (1 equiv.), (c) G3. | ||
Fluorescence analysis of G3 and WPA5·G3 complex
The optical spectral data of G3 and its WPA5·G3 complex in aqueous solution are collected in Table 1. The positions of the absorption and fluorescence bands of G3 are sensitive primarily to the polarity and the hydrogen bond acceptor ability of the local environment, quantified by the π* and β Kamlet–Taft solvatochromic parameters, respectively.21The absorption maximum of the dye shifts to lower wavelengths with increasing solvent polarity (negative solvatochromism), in accordance with the results of theoretical calculations.22 These showed that G3 is a charge transfer dye with a strongly polar S0 state, in which the positive charge is concentrated on the pyridinium moiety, and a less polar S1 state, where the charge is more evenly distributed on the pyridinium and aniline segments. Hydrogen bond acceptor, i.e. electron pair donor solvents stabilize the G3 solute by interacting with the positive charge of the ground as well as the excited state solute. The net effect is a bathochromic shift of the absorption band with increasing β parameter. The absorption band of G3 complexed by WPA5 – 515 nm vs. 448 nm measured in water – falls at a higher wavelength than in the solution spectra measured in most of solvents (the exception is the CH2Cl2 solution of the dye with λabs = 517 nm),23 demonstrating that the less polar excited state solute is stabilized strongly in the cavity of the WPA5 host.
Unlike the absorption band, the fluorescence band of G3 shows a bathochromic shift upon complexation, but this is also in accord with the lower polarity of the environment, as the fluorescence band of the dye shows a positive solvatochromism.
The spectacular enhancement of the fluorescence intensity upon complexation is a consequence of hindering the deactivation via non-emissive twisted intramolecular charge-transfer (TICT) states. The results of time-resolved fluorescence studies in low temperature ethanol solutions suggested that the planar excited-state of G3 formed via vertical excitation from the coplanar ground state molecule, easily relaxes to energetically close non-planar structures.24 Theoretical calculations showed that the non-planar species are formed presumably via the rotations of the aniline or pyridinium units around the single C–C bonds of the vinylene group, as the torsion of the dimethylamino group and the twisting around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond are energetically less favored.25 The correlation of the fluorescence lifetimes (τF) of G3 measured in water–glycerol mixtures with viscosity of the solvent,26 and the correlation of the τF values in polymer matrices with the elastic modulus of the medium27 are also consistent with the decisive role of a non-emissive TICT state of the dye in the deactivation process.
C double bond are energetically less favored.25 The correlation of the fluorescence lifetimes (τF) of G3 measured in water–glycerol mixtures with viscosity of the solvent,26 and the correlation of the τF values in polymer matrices with the elastic modulus of the medium27 are also consistent with the decisive role of a non-emissive TICT state of the dye in the deactivation process.
The picosecond decay kinetics of G3 in room temperature solutions can be mono-, bi- and triexponential, depending on the polarity and viscosity of the solvent, which can be interpreted in terms of an energy-level scheme with three sub-levels within the S1 state: a weakly polar planar ‘locally excited’ (LE), a strongly polar, also planar intramolecular (ICT) state, and the above TICT state.28 In strongly polar, low viscosity solvents, like water, the decay is fast and monoexponential, as the emission derives only from the LE state, the ICT state easily deactivates via dipole–dipole solute–solvent interactions. The TICT state may be emissive only in highly polar and viscous solvents, like glycerol, where it may contribute to the emission on the red edge of the fluorescence band. By a different model, describing the deactivation of the dye on the potential energy surface (PES),29 the TICT excited state of G3 can be considered a ‘sink’ region on the PES, and the relaxation of the molecule to the TICT state, following the excitation, is a coupled process involving contributions from the solvational, vibrational and torsional relaxations.
A similarly strong enhancement of fluorescence intensity was observed with the cucurbit[6]uril complex of G3,30 whereas its β-cyclodextrin31 and cucurbit[7]uril32 complexes showed a somewhat smaller effect.
The fluorescence lifetime of G3, τF, in aqueous solution is shorter than ∼20 ps,33 the temporal resolution of our TCSPC system. The decay of the WPA5·G3 complex was found much slower, and was best fitted by a biexponential function with two close time constants. The decay curves measured at different wavelengths on the rising and falling edges of the fluorescence band were found similar, their time constants and relative amplitudes were not sensitive to the wavelengths. This is illustrated in Fig. S6,† showing the time-resolved emission spectra constructed from the decay curves, which did not shift on the picoseconds time scale. This differs from the decay kinetics of the dye in various solvents, which change markedly with the wavelength, following the different phases of structural and solvational relaxation.28,29,33
Fluorescence indicator displacement (FID)
DMV (dimethylviologen or 4,4′-dimethylbipyridinium salts like paraquat) are known to bind strongly to WPA5via inclusion in the cavity,7 therefore by designing a threaded macrocycle-dye supramolecular system with modified fluorescence compared to the free dye, the competitor paraquat can be detected or measured by the displacement of the dye. The fluorescence intensity of the complex can be higher or lower than the free dye, hence the analyte exhibits turn-off or turn-on fluorescence response, respectively.34To our knowledge, only a few examples of pillararene-based receptors are used in FID methods, thus we speculated that our most sensitive system could be used for the detection of DMV analyte. The addition of DMV (diiodide salt) to the WPA5·G3 system (Fig. 4) caused the expected fluorescence quenching (turn-off sensing) due to the dethreading of the dye from the complex causing the regeneration of the almost non-fluorescent free state of G3 in water. The displacement, caused by the stronger binding of DMV can be easily observed by the color changes and the reduced emission under a handheld UV lamp.
 | ||
| Fig. 4 Absorption and fluorescence spectra of G3 (5.0 μM), WPA5 (15.0 μM) upon addition of DMV (0 μM to 27.4 μM) in water (excitation at 506 nm); inset: photographs showing colorimetric and fluorescent changes in aqueous solution (10.0 μM G3 and 30.0 μM WPA5 (left vial), 10.0 μM G3, 30.0 μM WPA5 and 30.0 μM DMV (left vial)). | ||
Fluorescence titration experiments proved our system to be very sensitive: a 60% reduction in fluorescence intensity can be observed by adding 15 μM of DMV to a solution of WPA5·G3 (5.0 μM dye and 15 μM host concentration) which is a significantly better result than the previously reported.11 From the absorption spectra in Fig. 4, K2 = (2.57 ± 0.4) × 106 M−1 was obtained for the equilibrium constant of the binding of DMV by the pillararene host by a least square fitting calculation (see ESI† for details). To characterize the sensitivity of this FID method, the fluorescence intensities of WPA5–G3-paraquat systems with fixed WPA5 and G3 initial concentrations and varying added paraquat concentrations were calculated from K1 and K2. As can be seen in Fig. S7,† the addition of paraquat in 0.2 μM concentration to a solution containing [G3]0 = 1 μM dye and [WPA5]0 = 3 μM pillararene, results in a ∼5% change of fluorescence intensity, that can be easily detected.
Conclusions
In summary, we have examined the interaction between different pyridinium-based stilbazolium salts and WPA5 as new host-guest systems where the interactions are detectable by optical spectroscopic methods. The most significant effect was observed in the case of G3 with remarkable turn-on fluorescence and color change thus WPA5·G3 system could be used in various supramolecular systems in the future. This was demonstrated by using it as a FID-based probe to detect paraquat in a highly sensitive manner. In addition, detailed description on the mechanism of fluorescence enhancement is provided.Acknowledgements
The authors are grateful for the financial support from the Hungarian Scientific Research Fund (OTKA, Grant no. K108752).Notes and references
- D. J. Cram, Angew. Chem., 1988, 100, 1041–1052 CrossRef CAS PubMed; D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 CrossRef PubMed; J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef PubMed; J.-M. Lehn, Angew. Chem., 1988, 100, 91–116 CrossRef PubMed; C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- P. J. Cragg and K. Sharma, Chem. Soc. Rev., 2012, 41, 597–607 RSC; T. Ogoshi, J. Inclusion Phenom. Macrocyclic Chem., 2012, 72, 247–262 CrossRef CAS PubMed; T. Ogoshi and T.-a. Yamagishi, J. Synth. Org. Chem., Jpn., 2012, 70, 842–851 CrossRef; M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef PubMed; H. Meier and D. Cao, Nachr. Chem., 2013, 61, 408–410 CrossRef PubMed; T. Ogoshi and T.-a. Yamagishi, Eur. J. Org. Chem., 2013, 2013, 2961–2975 CrossRef PubMed; H. Zhang and Y. Zhao, Chem.–Eur. J., 2013, 19, 16862–16879 CrossRef PubMed; N. L. Strutt, H. Zhang, S. T. Schneebeli and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2631–2642 CrossRef PubMed.
- T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76, 328–331 CrossRef CAS PubMed.
- T. Ogoshi, N. Ueshima, T. Akutsu, D. Yamafuji, T. Furuta, F. Sakakibara and T.-a. Yamagishi, Chem. Commun., 2014, 50, 5774–5777 RSC.
- T. Ogoshi, N. Ueshima, F. Sakakibara, T.-a. Yamagishi and T. Haino, Org. Lett., 2014, 16, 2896–2899 CrossRef CAS PubMed.
- T. Ogoshi, M. Hashizume, T.-a. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46, 3708–3710 RSC.
- Y. Ma, X. Ji, F. Xiang, X. Chi, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Chem. Commun., 2011, 47, 12340–12342 RSC.
- N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS PubMed.
- C. Han, F. Ma, Z. Zhang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef CAS PubMed; C. Li, Q. Xu, J. Li, Y. Feina and X. Jia, Org. Biomol. Chem., 2010, 8, 1568–1576 Search PubMed; C. Li, L. Zhao, J. Li, X. Ding, S. Chen, Q. Zhang, Y. Yu and X. Jia, Chem. Commun., 2010, 46, 9016–9018 RSC.
- P. Wang, Y. Yao and M. Xue, Chem. Commun., 2014, 50, 5064–5067 RSC.
- C. Li, J. Ma, L. Zhao, Y. Zhang, Y. Yu, X. Shu, J. Li and X. Jia, Chem. Commun., 2013, 49, 1924–1926 RSC.
- P. Wang, X. Yan and F. Huang, Chem. Commun., 2014, 50, 5017–5019 RSC.
- D. Li, D. Yu, Q. Zhang, S. Li, H. Zhou, J. Wu and Y. Tian, Dyes Pigm., 2013, 97, 278–285 CrossRef CAS PubMed; F. Hao, D. Zhu, J. Ma and L. Chai, Spectrochim. Acta, Part A, 2014, 123, 46–53 CrossRef PubMed.
- G. R. Rosania, J. W. Lee, L. Ding, H.-S. Yoon and Y.-T. Chang, J. Am. Chem. Soc., 2003, 125, 1130–1131 CrossRef CAS PubMed.
- J. N. Wilson, A. S. Brown, W. M. Babinchak, C. D. Ridge and J. D. Walls, Org. Biomol. Chem., 2012, 10, 8710–8719 CAS.
- C. W. Chan, T. F. Lai, C. M. Che and S. M. Peng, J. Am. Chem. Soc., 1993, 115, 11245–11253 CrossRef CAS; K. N. Koh, K. Araki, A. Ikeda, H. Otsuka and S. Shinkai, J. Am. Chem. Soc., 1996, 118, 755–758 CrossRef; G. R. Clemo and G. A. Swan, J. Chem. Soc., 1938, 1454–1455 RSC.
- R. R. Kothur, J. Hall, B. A. Patel, C. L. Leong, M. G. Boutelle and P. J. Cragg, Chem. Commun., 2014, 50, 852–854 RSC.
- B. Juskowiak and M. Chudak, Photochem. Photobiol., 2004, 79, 137–144 CrossRef CAS.
- A. Rei, M. B. Graham Hungerford, M. Isabel, C. Ferreira and P. Schellenberg, Int. J. Spectrosc., 2012, 2012 Search PubMed.
- F. Moyano, J. J. Silber and N. M. Correa, J. Colloid Interface Sci., 2008, 317, 332–345 CrossRef CAS PubMed.
- S. T. Abdel-Halim and M. K. Awad, J. Mol. Struct., 2009, 920, 332–341 CrossRef CAS PubMed.
- M. Panigrahi, S. Patel and B. K. Mishra, J. Mol. Liq., 2013, 177, 335–342 CrossRef CAS PubMed.
- B. Strehmel and W. Rettig, BIOMEDO, 1996, 1, 98–109 Search PubMed; B. Strehmel, H. Seifert and W. Rettig, J. Phys. Chem. B, 1997, 101, 2232–2243 CrossRef CAS.
- M. Dekhtyar and W. Rettig, J. Phys. Chem. A, 2007, 111, 2035–2039 CrossRef CAS PubMed.
- A.-Y. Jee, E. Bae and M. Lee, J. Chem. Phys., 2010, 133 Search PubMed.
- A.-Y. Jee and M. Lee, ChemPhysChem, 2010, 11, 793–795 CrossRef CAS PubMed.
- R. Ramadass and J. Bereiter-Hahn, J. Phys. Chem. B, 2007, 111, 7681–7690 CrossRef CAS PubMed.
- Y. Huang, T. Cheng, F. Li, C.-H. Huang, S. Wang, W. Huang and Q. Gong, J. Phys. Chem. B, 2002, 106, 10041–10050 CrossRef CAS.
- Z. Li, S. Sun, F. Liu, Y. Pang, J. Fan, F. Song and X. Peng, Dyes Pigm., 2012, 93, 1401–1407 CrossRef CAS PubMed.
- J. W. Park, S. Y. Lee and S. M. Kim, J. Photochem. Photobiol., A, 2005, 173, 271–278 CrossRef CAS PubMed.
- S. Sun, Y. Yuan, Z. Li, S. Zhang, H. Zhang and X. Peng, New J. Chem., 2014, 38, 3600–3605 RSC.
- A. Rei, G. Hungerford, M. Belsley, M. I. C. Ferreira and P. Schellenberg, Int. J. Spectrosc., 2012, 2012, 1–5 CrossRef PubMed.
- G. Ghale and W. M. Nau, Acc. Chem. Res., 2014, 47, 2150–2159 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods and additional spectroscopic information. See DOI: 10.1039/c4ra14809f |
| This journal is © The Royal Society of Chemistry 2015 |