
Necessity of moderate metal-support interaction in Ni/Al2O3 for syngas methanation at high temperatures
Abstract
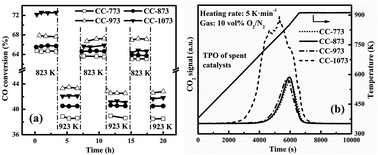
This article investigates the necessary metal-support interaction in the Ni–Mg–Zr–La/Al2O3 catalyst for high-temperature syngas methanation through varying the calcination and reduction temperatures during catalyst preparation. Atmospheric syngas methanation was conducted continuously but at 823 K and 923 K alternatively. Raising the calcination temperature increased the interaction between the nickel and alumina support but lowered the surface area of the catalyst. The catalyst calcined at the higher temperature such as 1073 K exhibited the highest initial activity in spite of its least active sites. However, only those catalysts with moderate interaction, such as that calcined at 973 K, showed stable activity of methanation above 823 K. Too small surface area of the catalyst calcined at too high temperatures, such as 1073 K, would lead to serious aggregation of nickel particles and thus cause carbon deposition during methanation. Varying the reduction temperature for the catalyst demonstrated that the excessive reduction of the nickel species such as that reduced at 1073 K almost removed all the nickel aluminates to cause the catalyst to have low activity, quick nickel aggregation and carbon deposition. This further justified the claim that moderate metal-support interaction is necessary to ensure the high activity and stability of the Ni/Al2O3 catalyst for syngas methanation at high temperatures.
 Please wait while we load your content...
Please wait while we load your content...

