
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular nitrogen in N-doped TiO2 nanoribbons†
C.
Bittencourt
*a,
M.
Rutar
bc,
P.
Umek
b,
A.
Mrzel
b,
K.
Vozel
b,
D.
Arčon
be,
K.
Henzler
d,
P.
Krüger
f and
P.
Guttmann
d
aChimie des Interactions Plasma Surface, CIRMAP, University of Mons, 23 Place du Parc, B-7000 Mons, Belgium. E-mail: carla.bittencourt@umons.ac.be
bJožef Stefan Institute, Jamova cesta 39, SI-1000 Ljubljana, Slovenia
cJožef Stefan International Postgraduate School, Jamova cesta 39, SI-1000 Ljubljana, Slovenia
dHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Institute for Soft Matter and Functional Materials, Albert-Einstein-Str. 15, D-12489 Berlin, Germany
eFaculty of Mathematics and Physics, University of Ljubljana, Jadranska cesta 19, SI-1000 Ljubljana, Slovenia
fGraduate School of Advanced Integration Science, Chiba University, Chiba 263-8522, Japan
First published on 20th February 2015
Abstract
The nitrogen doping of TiO2 nanoribbons during the thermal transformation of hydrogen titanate nanoribbons (HTiNRs) between 400 and 650 °C in a dynamic ammonia atmosphere was investigated using X-ray photoelectron spectroscopy (XPS), transmission X-ray microscopy combined with near-edge X-ray absorption fine structure spectroscopy (NEXAFS-TXM), X-ray diffraction (XRD) and electron paramagnetic resonance measurements (EPR). Comprehensive structural characterizations have revealed that for a calcination temperature of 400 °C, the HTiNRs transform into pure monoclinic TiO2 β-phase (TiO2-B) whereas at higher calcination temperatures (580 and 650 °C) a mixture of TiO2-B and anatase is obtained. XPS and EPR results clearly reveal the nitrogen doping of TiO2 nanoribbons and that, depending on the calcination temperature, nitrogen atoms occupy interstitial and substitutional sites. Moreover, in samples calcined at 580 and 650 °C the presence of N2-like species in the HTiNRs was detected by NEXAFS-TXM. These species are trapped in the HTiNRs structure. EPR measurements upon light illumination have disclosed the generation of photoexcited states which implies that nitrogen has an important effect on the electronic structure of N-doped TiO2.
Introduction
Nitrogen doping titanium dioxide (TiO2), with N at substitutional sites has been reported to be indispensable for enhancing the use of TiO2 as a visible-light photocatalytic material.1 The main effect of the N-doping is the narrowing of the energy band gap of TiO2 due to the mixing of N 2p and O 2p states.2 Additionally, an isolated narrow band responsible for the visible light photoactivity is formed above the valence band.2 In substitutional N-doping, the N atom bonds to three Ti atoms replacing a lattice O atom in TiO2. Otherwise, N atoms can also occupy interstitial sites where N is bound to one or more O atoms thus creating paramagnetic centres. The electronic properties of N-doped titania, the corresponding bonding configuration, and the oxidation states of nitrogen in the lattice are yet to be unambiguously determined.3–5 Recent reports based on density functional theory (DFT) calculations and are consistent with both substitutional and interstitial nitrogen doping,6–9 where interstitial doping is slightly lower in energy especially for surface states with low concentration of oxygen vacancies.3 For further improving the photocatalytic performance of titania, it is thus critical to identify the local bonding configurations around N atoms at the atomic level.Nitrogen doping into the TiO2 lattice is rather challenging as N atoms must be accommodated geometrically and electronically.2,10–12 To address the fundamental issues discussed above we explored a chemical route and report here a comprehensive investigation of N-doped TiO2 nanoribbons. As a precursor material for N-doped TiO2 nanoribbons we used hydrogen titanate nanoribbons (HTiNRs) because upon heating HTiNRs easily transform first to the monoclinic TiO2 (ref. 13) β-phase (TiO2-B) and then to anatase (tetragonal phase) while retaining the morphology of the parent nanostructure intact.14 In the used chemical route, the precursor HTiNRs were calcined at different temperatures for different times in an NH3(g)/Ar(g) flow (Table 1). When calcined at 400 °C the HTiNRs successfully converted to TiO2-B, whereas calcination at 580 °C and 650 °C yielded in a mixture of two TiO2 phases. Morphology probing transmission electron microscopy (TEM) and structure sensitive X-ray diffraction (XRD) techniques were employed to extract critical local and global bonding information. The nitrogen chemical environment was inspected with X-ray photoelectron spectroscopy (XPS), while its bond geometry was probed by transmission X-ray microscopy combined with near-edge X-ray absorption fine structure spectroscopy (NEXAFS-TXM). The nitrogen chemical environment (i.e., substitutional or interstitial) was found to depend on the calcination temperature. In N-doped TiO2 nanoribbons calcined at 580 °C and 650 °C, besides the expected covalently bonded nitrogen, surprisingly, also N2-like species were detected through N 1s XPS and N K-edge NEXAFS. The present results provide a coherent description of N-sites in TiO2 nanostructures and may thus facilitate further improvements in their photocatalytic performance.
| Sample | T (°C) | t (h) | N content* (wt%) | Phase composition |
|---|---|---|---|---|
| N400-6h | 400 | 6 | 0.8 | TiO2-B |
| N580-6h | 580 | 6 | 1.3 | Anatase, TiO2-B |
| N650-4h | 650 | 4 | 2.8 | Anatase, TiO2-B, traces of titanium oxynitride phase |
Experimental
Synthesis of N-doped TiO2 nanoribbons
N-doped TiO2 nanoribbons were synthesized from hydrogen titanate nanoribbons (HTiNRs) by calcination in a dynamic NH3(g)/Ar(g) atmosphere. The reaction was carried out in a horizontal furnace, which was purged with a mixture of NH3(g)/Ar(g). The NH3(g)/Ar(g) flow ratio was set to 30 mL min−1/10 mL min−1, respectively. In a typical experiment 225 mg of HTiNRs, prepared from sodium trititanate nanoribbons15 by an ion-exchange process,14 is weighted on a quartz boat, placed into the hot zone of the furnace and heated at a ramp rate of 7 °C min−1 to a selected temperature. The samples were calcined at the selected temperature for four or six hours. The experimental conditions for the transformation of HTiNRs to TiO2 nanoribbons, nitrogen content and phase composition are summarized in Table 1.Characterization techniques
The morphology of the N-doped TiO2 nanoribbon samples was investigated with a field emission scanning (FE-SEM, Jeol, 7600F) and a transmission (TEM, Jeol 2100) electron microscopes.The phase analysis was performed on the cut surface by X-ray powder diffraction (XRD) using a diffractometer with Cu Kα radiation (λ = 1.5406 Å) and a Sol-X energy-dispersive detector (Endeavor D4, Bruker AXS, Karlsruhe, Germany).
The XPS measurements were performed in a VERSAPROBE PHI 5000 from Physical Electronics, equipped with a Monochromatic Al Kα X-ray source. The energy resolution was 0.7 eV. For the compensation of built up charge on the sample surface during the measurements a dual beam charge neutralization composed of an electron gun (∼1 eV) and an Argon ion gun (≤10 eV) was used. The samples for XPS measurements were prepared by pressing the sample into a pellet. A conductive double face tape was used to attach the pellet to a sample holder.
The near edge X-ray adsorption fine structure (NEXAFS) spectra were recorded with the TXM (transmission X-ray microscope) end-station installed at the undulator beamline U41-XM at the electron storage ring BESSY II, Helmholtz-Zentrum Berlin (HZB).16 The spectra were recorded at room temperature in a transmission mode by taking a sequence of images over a range of photon energies covering the investigated absorption edges with E/ΔE ≥ 4500. The exit slit of the monochromator was set to 5 μm, which corresponds to a calculated spectral resolution of E/ΔE = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The data analysis was accomplished by using aXis2000.17 The NEXAFS spectra were normalized by using the signal intensity in the proximity of the sample to correct for variations of the photon flux with photon energy (hν) and acquisition time. For comparison and calibration molecular nitrogen N2(g) was analyzed using Nitrogen 5.0 with 99.999% purity (Air Liquide).
000. The data analysis was accomplished by using aXis2000.17 The NEXAFS spectra were normalized by using the signal intensity in the proximity of the sample to correct for variations of the photon flux with photon energy (hν) and acquisition time. For comparison and calibration molecular nitrogen N2(g) was analyzed using Nitrogen 5.0 with 99.999% purity (Air Liquide).
For the TEM and NEXAFS-TXM analyses the samples were sonically dispersed in ethanol and a drop of the solution was deposited onto a lacey carbon film supported by a copper grid. Note that due to electron-beam damage effects on the N-doped TiO2 nanostructures, different grids were used for the TEM and for the NEXAFS-TXM analysis.
The EPR measurements were conducted using a commercial Bruker E580 spectrometer equipped with an Oxford Cryogenics He flow cryostat that has an optical window. The spectra were measured by recording the intensity of the echo signal as a function of magnetic field. A Hahn-echo pulse sequence (π/2–τ–π–τ–echo) together with appropriate phase cycling has been used. The length of π/2 pulse was set to 20 ns and the delay between the two pulses was τ = 200 ns. The sample light-illumination was performed in situ at 35 K with a standard halogen lamp. A sample for EPR measurements was sealed under dynamic vacuum in a standard Suprasil quartz EPR tube.
Results and discussion
The typical morphology of the nanostructures prepared by calcination in the NH3(g)/Ar(g) flow, presented in Fig. 1, show that transformation from hydrogen titanate to N-doped TiO2 does not affect the nanoribbon morphology. The nanoribbons reach a length of several micrometres with diameters in the range of 30 to 250 nm and are thus comparable to the starting hydrogen titanate nanoribbon (HTiNRs) sample.18 TEM image of N580-6h (Fig. S1A†) reveals a crystalline nature of the N-doped TiO2 nanoribbons. However, in striking contrast to the parent material, a close inspection of the nanoribbons' surface reveals their porous structure (Fig. S1B†) during the transformation of HTINRs to N-doped TiO2 nanoribbons14 in the NH3(g)/Ar(g) atmosphere. The porous structure is characteristic of TiO2–B nanoribbons and with further transformation to anatase disappears as we reported in ref. 14. | ||
| Fig. 1 SEM images of (A) starting hydrogen titanate nanoribbons and N-doped TiO2 nanoribbons prepared from hydrogen titanate nanoribbons by calcination in an NH3(g)/Ar(g) flow for 6 h at (B) 400 °C, (C) 580 °C and (D) 650 °C. | ||
The crystal structure of samples treated at different calcination temperatures was examined by powder XRD (Fig. 2). The XRD pattern of the sample calcined at 400 °C reveals only the presence of the TiO2-B phase (JCPDS no. 35-0088), whereas the samples treated at 580 °C and 650 °C come as a mixture of TiO2-B and anatase phases.13,19 The amount of anatase in the samples increases with increasing calcination temperature up to 650 °C. At this temperature TiO2 already starts to transform to titanium oxynitride as indicated with the appearance of a new diffraction peak at 2θ ∼ 43°.20
 | ||
| Fig. 2 XRD patterns of N-doped TiO2 nanoribbons which were obtained by calcination of hydrogen titanate nanoribbons in an NH3(g)/Ar(g) flow at 400 °C (6 h), 580 °C (6 h) and 650 °C (4 h). Anatase peaks are labelled with asterisks (*), TiO2-B peaks with triangles (▲), and titanium oxynitride phase with a diamond (♦). Two vertical lines positioned at (110) and (020) diffractions of TiO2-B are added for easier notion of slightly different positions of (110) diffraction of TiO2-B and (101) diffraction of anatase, and (020) diffraction of TiO2-B and (200) diffraction of anatase. | ||
The XPS analysis of the samples confirmed the presence of nitrogen in all calcined samples (Table 1). The relative content of nitrogen increases with the calcination temperature in agreement with data published in the literature.21–23 The samples with different nitrogen contents have colours that range from white over light yellow to light green. The higher the nitrogen content, the more intense is the colour of the sample. The XPS Ti 2p spectra show a Ti 2p3/2 binding energy of 458.7 eV with a splitting of 5.7 eV between the doublets, characteristic of titanium in the oxidation state 4+ (Fig. 3A, S2 and S3†).3,24 The absence of peaks corresponding to titanium in lower oxidation states in the XPS spectra of N400-6h and N580-6h suggests that there is no significant net electron transfer to titanium atoms from oxygen vacancy formation.3 Conversely, in the spectrum recorded on the sample calcined at 650 °C (N650-4h) we observed a low intensity shoulder at low binding energies characteristic of the photoelectrons generated in titanium species having lower oxidation states (Ti+3 at 457.3 eV and Ti+2 455.5 eV)25 (Fig. S3†).
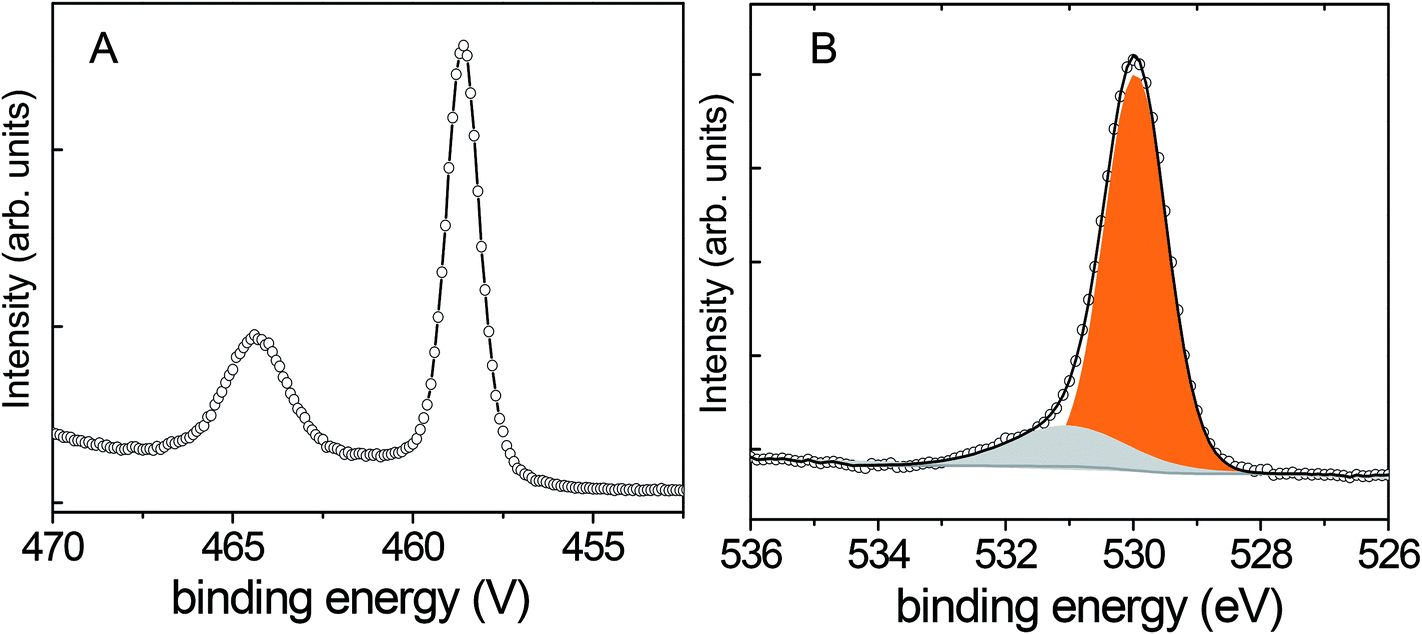 | ||
| Fig. 3 XPS spectra of N580-6h: (A) Ti 2p and (B) O 1s. | ||
Fig. 3B shows the typical O 1s XPS spectrum recorded on the studied samples (Fig. S2B and S3B†). It exhibits a major peak centred at 530.0 eV, which is characteristic of transition metal oxides. The broad peak centred at 531.2 eV was attributed to hydroxyl groups and carbon compounds contamination at the TiO2 surface.2,26 Most likely the carbon contamination originates from the preparation of HTiNRs14 where ethanol was used for the final rinsing of HTiNRs after the ion exchange.
The N 1s XPS peaks recorded on N580-6h and N650-4h, show four components whereas in the N1s peak of N400-6h only three components are observed (Fig. 4). The component with binding energy at 396.1 eV is assigned to O–Ti–N linkage in the lattice, i.e. lattice oxygen atoms are replaced by nitrogen atoms (substitutional N-doping). The relative intensity of this component and the total amount of nitrogen incorporated increased with increasing calcination temperature in accordance with the results reported by Zhang et al. on the N-doping of on titania nanoparticles prepared by a sol–gel technique.21 The component at 396.7 eV was assigned to carbon contamination26 as observed in the O 1s XPS spectra, while the component at 400.1 eV to nitrogen species bonded to oxygen sites (N–O–Ti linkage); i.e. an interstitial N-doping mode formed by N-atoms bonded to one or more lattice oxygen atoms.2 The contribution of the component associated to photoelectrons emitted from N atoms in O–Ti–N linkages to the N 1s XPS spectrum increases at the expense of the other three components as the calcination temperature increases. DFT calculations showed that adsorbed and interstitial nitrogen atoms have a tendency to bond to oxygen vacancy sites and accept electrons,6 explaining the high intensity of the component associated to N atoms in the O–Ti–N linkage. The highest binding energy peak at 403.7 eV has been ascribed to molecular N2.24,27 This binding energy is significantly lower than for gas phase N2 molecules (409.9 eV), indicating that photocreated core holes maybe screened by neighbouring atoms as was observed in physisorbed N2 molecules on various surfaces.28 In order to investigate the presence of N2 molecules in the N-doped TiO2 nanoribbon samples (N580-6h and N650-4h), we next studied the Ti L-edge and the N K-edge using the NEXAFS-TXM, which allows recording NEXAFS spectra from isolated nanoribbons or selected regions of the sample.16 The features of these edges are sensitive to the local symmetry and ligand coordination, providing detailed information on crystal structure, oxidation states of solid samples and vibrational fine-structure of gas molecules.
 | ||
| Fig. 4 Comparison of the N 1s XPS spectra for the N-doped TiO2 NRs derived from hydrogen titanate nanoribbons calcined in an NH3(g)/Ar(g) flow at (A) 400, (B) 580, and (C) 650 °C. The peak at 403.7 is assigned to molecular N2. The peak appearing at 396.3 eV is attributed to the O–Ti–N linkage (substituional N-doping), the relative intensity of this peak increases at the expense of the N2 and N–O–Ti linkage (interstitial) as the calcination temperature increases. | ||
The TXM-NEXAFS spectra were recorded on a region of interest of an isolated nanoribbon (Fig. S4†). The Ti L-edge spectrum of the N-doped TiO2 nanoribbons contains two sets of doublets (Fig. S5,† column A), Ti L3 at lower energy and Ti L2 at higher energy. The two doublets originate primarily from electronic transitions from the Ti 2p3/2 and Ti 2p1/2 levels to Ti 3d orbitals, which split into t2g and eg levels in the approximately octahedral symmetry such as in the anatase TiO2 phase.29 The features of these Ti L-edge indicate that the coordination environment of the titanium atoms in the N-doped TiO2 samples corresponds to Ti(IV) in a tetragonal structure as suggested by the XPS measurements.16
The N K-edge spectra recorded on N580-6h and N650-4h show a very sharp peak at 401 eV that is not present in the spectrum recorded on N400-6h (Fig. 5). To examine this more carefully, we recorded detailed spectra in the 399–403 eV photon energy range with step energy of 0.05 eV and compared with the spectrum recorded on a standard sample of molecular nitrogen (Fig. 6). In both cases we observed five well-resolved peaks that are characteristic of free N2 molecules.24,30,31 The peak at 400.8 eV of the gas phase N2 molecules was assigned to a transition from the N 1s to π* orbital and the fine feature is a hallmark of N2 molecule due to its vibrational structure.32 Therefore, the observed fine structure in the N K-edge undoubtedly proves the presence of N2 molecules in the N-doped TiO2 nanoribbons. This is further supported by the observation that the energy position of the 5 components of the peak, their area ratio, and energy separation are similar to those of gas phase N2 molecule. This means that N–N bond order of N2 in the N-doped TiO2 nanoribbons is comparable to that of gas phase N2 molecule and the interaction of N2 with the N-doped TiO2 NRs can be considered as very weak.30 Therefore, the N 1s XPS and the N K-edge NEXAFS spectra support the idea that nitrogen atoms trapped in N-doped TiO2 nanoribbons may combine to form N2 molecules evolving into the gas phase upon heating at elevated temperatures. This behaviour agrees well with reported DFT calculations which show that nitrogen embedded in TiO2 is unstable energetically and spontaneously forms trapped or gaseous N2.3 Therefore, the XPS and the NEXAFS analyses suggest that for the doping parameters used, the incorporated nitrogen atoms remain in the oxide structure as substitutional or interstitial sites and do not segregate into the ordered titanium oxynitride phase for calcination temperature in the range of 400 to 580 °C; segregation was observed at 650 °C.
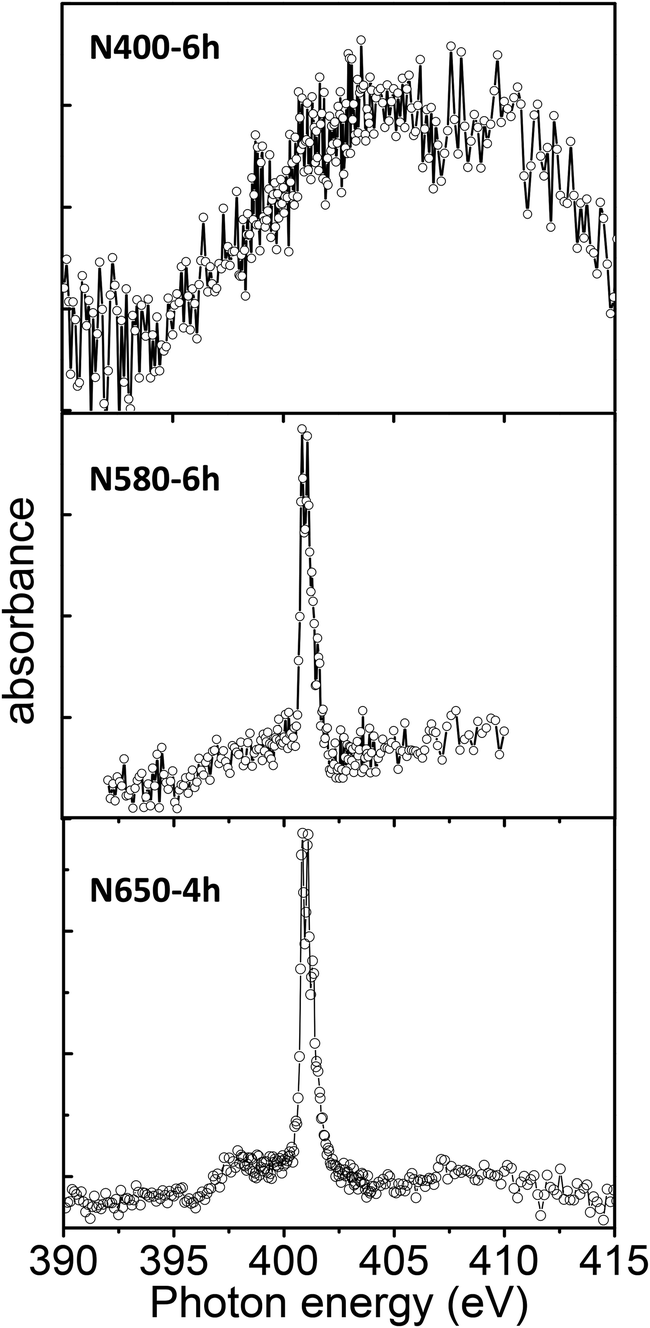 | ||
| Fig. 5 NEXAFS-TXM spectra of the N K-edge of the N-doped TiO2 nanoribbons recorded with an energy step of 0.07 eV. | ||
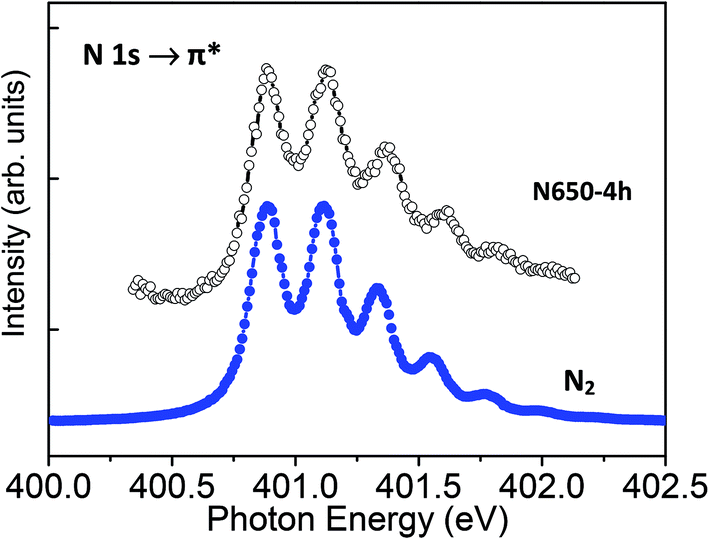 | ||
| Fig. 6 Comparison of high-energy resolution NEXAFS-TXM N K-edge spectra (step energy 0.05 eV) recorded on 650-4h and on N2 gas. The characteristic vibrational structures in the spectrum of the N-doped TiO2 nanoribbons indicate the presence of N2 molecules. | ||
The formation of paramagnetic NOx defects and the possible changes in the electronic structure after nitrogen doping prompted us to carry out electron paramagnetic resonance (EPR) measurements. EPR has proved to be a very powerful tool for the investigation of local properties of various paramagnetic centres in TiOx nanostructures.18,33–37 Moreover, it can also distinguish between different NOx centers through the values of electronic g-factors and hyperfine coupling tensors A.18,38,39 The X-band EPR spectrum of N580-6h sample measured at 35 K with the echo-detected field-sweep technique is shown in Fig. 7A. Clearly, at least three different signals can be recognised. The broad component at high fields can be simulated with the uniaxial g-factor anisotropy yielding g⊥ = 1.907 and g‖ = 1.939. These eigenvalues match those expected for the Ti3+ surface sites, (Ti3+)S.40 The large homogenous broadening (ΔB1/2 = 4.7 mT) used in the simulation for this component is consistent with the strains and local inhomogeneities associated with such surface states and the fit could be improved if a distribution of g-factor values is considered. Much narrower homogenous broadening (ΔB1/2 = 0.9 mT) is characteristic for the second component with typical g⊥ = 1.974 and g|| = 1.986, also obtained from the powder lineshape fit. These values allow us to assign this signal to the bulk Ti3+ sites, (Ti3+)B.41 Finally, the fitting of the low-field signal at g = 2.0054 with two characteristic powder hyperfine satellite lines flanking the sharp peak yield the hyperfine coupling values of A⊥ = 18 MHz and A|| = 94 MHz. Both, g-factor and hyperfine coupling values are characteristic values for NO paramagnetic centres18 and provide independent support for the formation of N–O–Ti linkage. The EPR data fully corroborates the results of the XPS study (Fig. 4) and thus unambiguously prove the incorporation of N into the TiO2 lattice.
 | ||
| Fig. 7 X-band electron paramagnetic resonance spectra of N580-6h measured at 35 K with an echo-detected field-sweep technique (grey shaded area) before (A) and after (B) illumination with light. The spectra fit to the sum of three different signals (solid red line) ascribed to paramagnetic NO and to bulk (Ti3+)B and surface (Ti3+)S sites, respectively. | ||
Additionally, since the nitrogen doping is also known to enhance visible light photoactivity in TiO2, we measured EPR spectra of illuminated samples. Interestingly, different components of the EPR spectrum respond differently to light illumination. The spectrum measured at 35 K after illumination (Fig. 7B) still contains the same NO, (Ti3+)B, and (Ti3+)S signals as their respective g-factors and hyperfine coupling constants coincide with those before the illumination (Fig. 7A). However, while the signal assigned to surface Ti3+ sites remains almost the same, the signals of NO and bulk Ti3+ centres double in intensity after 10 min of illumination. This finding thus implies that photoexcited charges preferentially trap in the bulk of the nanoribbons where they form stable paramagnetic centres.
Conclusions
We successfully prepared N-doped TiO2 nanoribbons using hydrogen titanate nanoribbons as a precursor by a thermal treatment in a dynamic ammonia atmosphere. Two types of nitrogen species with bonding energies of 396.1 eV and 400.1 eV were observed by X-ray photoelectron spectroscopy. The two nitrogen species can be assigned to substitutional nitrogen doping and nitrogen bonded to oxygen sites (interstitial), respectively. The amount of the substitutional nitrogen increases with the increasing calcination temperature. The NEXAFS-TXM studies show that local bonding environments of oxygen and titanium atoms in N-doped TiO2 nanoribbons are similar to the undoped TiO2. Depending on the calcination temperature N2 molecules can be formed and remain weakly trapped in the N-doped TiO2 structure. The changes observed in the EPR spectrum upon light illumination indicate that photoexcited charges preferentially trap in the bulk of the nanoribbons where they form stable paramagnetic centres.Acknowledgements
This work is financially supported by the Helmholtz-Zentrum Berlin für Materialien und Energie GmbH. The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 312284. We thank HZB for the allocation of synchrotron radiation beamtime at the undulator beamline U41-TXM at the BESSY II electron storage ring, Berlin. C. Bittencourt is Research Associate of the National Funds for Scientific Research (FRS-FNRS, Belgium).References
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- H. H. Ou, S. L. Lo and C. H. Liao, J. Phys. Chem. C, 2011, 115, 4000–4007 CAS.
- H. Chen, A. Nambu, W. Wen, J. Graciani, Z. Zhong, J. C. Hanson, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2007, 111, 1366–1372 CAS.
- M. Zukalova, J. Prochazka, Z. Bastl, J. Duchoslav, L. Rubacek, D. Havlicek and L. Kavan, Chem. Mater., 2010, 22, 4045–4055 CrossRef CAS.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96, 026103 CrossRef.
- A. Nambu, J. Graciani, J. A. Rodriguez, Q. Wu, E. Fujita and J. F. Sanz, J. Chem. Phys., 2006, 125, 094706 CrossRef CAS PubMed.
- C. Di Valentin, G. Pacchioni and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 085116 CrossRef.
- C. Di Valentin, A. Tilocca, A. Selloni, T. J. Beck, A. Klust, M. Batzill, Y. Losovyj and U. Diebold, J. Am. Chem. Soc., 2005, 127, 9895–9903 CrossRef CAS PubMed.
- C. Di Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414–11419 CrossRef CAS PubMed.
- X. Sun and Y. Li, Chem.–Eur. J., 2003, 9, 2229–2238 CrossRef CAS PubMed.
- H. H. Ou, C. H. Liao, Y. H. Liou, J. H. Hong and S. L. Lo, Environ. Sci. Technol., 2008, 42, 4507–4512 CrossRef CAS.
- H. H. Ou and S. L. Lo, Sep. Purif. Technol., 2007, 58, 179–191 CrossRef CAS PubMed.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 2286–2288 CrossRef CAS PubMed.
- P. Umek, C. Bittencourt, P. Guttmann, A. Gloter, S. D. Škapin and D. Arčon, J. Phys. Chem. C, 2014, 118, 21250–21257 CAS.
- P. Umek, R. C. Korošec, B. Jančar, R. Dominko and D. Arčon, J. Nanosci. Nanotechnol., 2007, 7, 3502–3508 CrossRef CAS PubMed.
- P. Guttmann, C. Bittencourt, S. Rehbein, P. Umek, X. Ke, G. Van Tendeloo, C. P. Ewels and G. Schneider, Nat. Photonics, 2012, 6, 25–29 CrossRef CAS.
- A. P. Hitchock, http://unicorn.mcmaster.ca/aXis2000.html.
- P. Umek, P. Cevc, A. Jesih, A. Gloter, C. P. Ewels and D. Arčon, Chem. Mater., 2005, 17, 5945–5950 CrossRef CAS.
- Y. V. Kolen'ko, K. A. Kovnir, A. I. Gavrilov, A. V. Garshev, J. Frantti, O. I. Lebedev, B. R. Churagulov, G. Van Tendeloo and M. Yoshimura, J. Phys. Chem. B, 2006, 110, 4030–4038 CrossRef PubMed.
- M. Zukalova, J. Prochazka, Z. Bastl, J. Duchoslav, L. Rubacek, D. Havlicek and L. Kavan, Chem. Mater., 2010, 22, 4045–4055 CrossRef CAS.
- Z. Zhang, X. Wang, J. Long, Q. Gu, Z. Ding and X. Fu, J. Catal., 2010, 276, 201–214 CrossRef CAS PubMed.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. Hong, A. Manivannan, M. Zhi, M. Li and N. Wu, J. Am. Chem. Soc., 2009, 131, 12290–12297 CrossRef CAS PubMed.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS.
- Y. Chung, J. C. Lee and H. J. Shin, Appl. Phys. Lett., 2005, 86, 022901–022903 CrossRef PubMed.
- C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr, NIST Standard Reference Database, Version 3.4, 2003, http:/srdata.nist.gov/xps/ Search PubMed.
- M. Grandcolas and J. Ye, J. Ceram. Process. Res., 2012, 13, 65–70 Search PubMed.
- P. Hoffmann and C. Pettenkofer, Physica Status Solidi (B) Basic Research, 2011, 248, 327–333 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer, 2nd edn, 2003 Search PubMed.
- C. Bittencourt, P. Krüger, M. J. Lagos, X. Ke, G. Van Tendeloo, C. Ewels, P. Umek and P. Guttmann, Beilstein J. Nanotechnol., 2012, 3, 789–797 CrossRef PubMed.
- M. Petravic, Q. Gao, D. Llewellyn, P. N. K. Deenapanray, D. Macdonald and C. Crotti, Chem. Phys. Lett., 2006, 425, 262–266 CrossRef CAS PubMed.
- J. Zhou, J. Wang, H. Liu, M. N. Banis, X. Sun and T. K. Sham, J. Phys. Chem. Lett., 2010, 1, 1709–1713 CrossRef CAS.
- J. Zhou, J. Wang, H. Liu, M. N. Banis, X. Sun and T. K. Sham, J. Phys. Chem. Lett., 2010, 1, 1709–1713 CrossRef CAS.
- P. Umek, M. Pregelj, A. Gloter, P. Cevc, Z. Jaglicic, M. Čeh, U. Pirnat and D. Arčon, J. Phys. Chem. C, 2008, 112, 15311–15319 CAS.
- P. Umek, C. Bittencourt, A. Gloter, R. Dominko, Z. Jaglicic, P. Cevc and D. Arčon, J. Phys. Chem. C, 2012, 116, 11357–11363 CAS.
- Z. V. Saponjic, N. M. Dimitrijevic, O. G. Poluektov, L. X. Chen, E. Wasinger, U. Welp, D. M. Tiede, X. Zuo and T. Rajh, J. Phys. Chem. B, 2006, 110, 25441–25450 CrossRef CAS PubMed.
- P. Szirmai, E. Horváth, B. Náfrádi, Z. Micković‡, R. Smajda, D. M. Djokić‡, K. Schenk, L. Fórró and A. Magrez, J. Phys. Chem. C, 2013, 117, 697–702 CAS.
- R. Arroyo, G. Cardoba, J. Padilla and V. H. Lara, Mater. Lett., 2002, 54, 397–402 CrossRef CAS.
- D. Biglino, H. Li, R. Erickson, A. Lund, H. Yahiro and M. Shiotani, Phys. Chem. Chem. Phys., 1999, 1, 2887–2896 RSC.
- H. Yahiro, A. Lund and M. Shiotani, Spectrochim. Acta, Part A, 2004, 60, 1267–1278 CrossRef PubMed.
- R. F. Howe and M. Grätzel, J. Phys. Chem., 1987, 91, 3906–3909 CrossRef CAS.
- R. F. Howe and M. Grätzel, J. Phys. Chem., 1985, 89, 4495–4499 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14410d |
| This journal is © The Royal Society of Chemistry 2015 |