
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-state NMR studies of nucleic acid components
Martin Dračínský
*a and
Paul Hodgkinson
b
aInstitute of Organic Chemistry and Biochemistry, Flemingovo nám. 2, 16610, Prague, Czech Republic. E-mail: dracinsky@uochb.cas.cz
bDepartment of Chemistry, Durham University, South Road, DH1 3LE, Durham, UK
First published on 9th January 2015
Abstract
Recent applications of solid-state NMR to the characterisation of nucleic acid systems are reviewed. Developments in NMR methodology and DFT-based first-principles calculations have led to the emergence of “NMR crystallography”, where solid-state NMR provides information on local structure and dynamics that complements information on periodic ordering and overall structure provided by traditional diffraction crystallography. The solid-state NMR is shown to provide direct information on hydrogen-bonding arrangements, metal ion interactions and local molecular dynamics that is difficult to obtain by other techniques, including solution-state NMR.
 Martin Dračínský | Martin Dračínský obtained his PhD in organic chemistry from Charles University in Prague in 2006. He is currently working as a researcher at Czech Academy of Sciences. His research deals with experimental solution and solid-state NMR spectroscopy applied mostly to modified nucleic acid components, theoretical prediction of NMR parameters (including vibrational and solvent effects) and classical and DFT molecular dynamics simulations. |
 Paul Hodgkinson | Paul Hodgkinson obtained his PhD in physical chemistry from Oxford University in 1995, and, following postdoc periods as UC Berkeley and the ENS de Lyon, has been working at Durham University's Department of Chemistry since 1998, where he is now a Reader in Magnetic Resonance. His research group works in both on the fundamental principles of solid-state NMR as well as applications to chemical problems, focussing on the use of quantum calculation to enable the “NMR crystallography” of molecular organic solids. |
Introduction
Nucleotides, oligonucleotides and nucleic acids are fascinating molecules that are responsible for many cellular processes such as the storage of genetic information, catalysis, metabolic regulation and energy supply.1 Furthermore, chemical modifications of the components of nucleic acids open up new ways of fighting many diseases, such as AIDS or cancer.2,3At first sight, using NMR to study solid nucleic acid components may seem somewhat perverse. Lack of motional averaging in the solid state means that NMR spectra of solids generally exhibit significantly poorer resolution than their solution-state counterparts, and it is generally the behaviour of nucleic acids in in vivo conditions rather than as solids that is of most biological interest. However, the overall re-orientational dynamics of molecules in solution significantly complicates the interpretation of solution-state NMR results. Moreover, the polyanionic nature of nuclei acids means that their solid forms are usually well hydrated and the local environments (which NMR probes) are, as a result, not too dissimilar from in vivo conditions. Solid-state NMR (SS-NMR) is now widely used as complement to classical diffraction methods of characterising solid materials, with Bragg diffraction providing the overall long-range structure and NMR providing information on aspects, such as hydrogen atom positioning and dynamics, that are difficult to characterise by diffraction. This field is commonly termed “NMR crystallography”, although it is important to note that NMR techniques are not limited to crystalline solids. Solid-state NMR has the great virtue that chemical shifts can be determined for solid materials of known structure, permitting the correlation of the NMR parameters directly with structural features.4,5 Solid-state NMR is thus a valuable tool for nucleoside and nucleotide structural studies since it provides the means to acquire spectra corresponding to single conformations, in contrast to solution NMR methods.
The fundamental concepts of solid-state NMR have been extensively described,6–8 and only summarised very briefly here. The various interactions of NMR are fundamentally anisotropic with respect to the strong magnetic field applied during the NMR experiment. In the solution state, however, fast molecular tumbling means that only the isotropic average of the interactions is observed via the NMR spectrum. Interactions with no isotropic component, such as the direct dipolar interaction between nuclear magnetic moments, and the quadrupolar interaction (due to coupling between nuclear electric quadrupole moments and local electric field gradients) have no effect on the spectrum, leaving just the isotropic components of the chemical shift and indirect (or J) coupling. This leads to sharp well-resolved solution-state spectra for spin-1/2 nuclei, such as 1H and 13C. This same molecular tumbling is an efficient driver of relaxation processes. This can be an asset, returning the spin states quickly to equilibrium and allowing the NMR experiment to be repeated quickly, but it can be problematic for nuclei with a significant electric quadrupole moment; rapid quadrupolar relaxation will typically lead to broad and unresolved lines for potentially useful nuclei such as 23Na. In contrast, the lack of extensive dynamics in the solid state means that NMR spectra of solid materials are strongly broadened by the anisotropic interactions. This broadening can be significantly reduced by the use of “magic-angle spinning” (MAS), which averages out, at least to first order, the anisotropic components of the NMR interactions. The extensive nature of dipolar coupling between 1H nuclei means that 1H NMR spectra of solids are still relatively poorly resolved, even at the fastest MAS spinning rates currently available (about 100 kHz), and so the initial nucleus of choice for organic systems is generally 13C. In contrast, specialised NMR techniques, such as the Multiple Quantum MAS (MQMAS) experiment, often allow better site resolution for nuclei such as 23Na to be obtained in the solid compared to the solution state. Similarly 2H NMR is a powerful technique for studying dynamics since the deuterium quadrupolar coupling is both small enough to be readily measured and highly sensitive to local dynamic processes. Dynamics of nucleic acids has been studied by 2H NMR for more than 30 years; early studies were reviewed in 1991.9 In contrast to other techniques (solution NMR, X-ray), solid-state NMR can probe motions with a broad range of time scales. Other specialised experiments, most notably REDOR10 experiments as discussed below, have been developed that allow dipolar couplings between pairs of nuclear spins to be measured directly. These provide more robust estimates of internuclear distances than the indirect estimation of dipolar couplings from “nuclear Overhauser effects” in the solution state.
In addition to methodological advances, the development of “NMR crystallography” has been driven by the availability of DFT calculations that allow NMR properties to be calculated efficiently.11,12 Such quantum calculations provide a direct link between structure and NMR observables; rather than the NMR spectrum simply being used as a fingerprint of a particular solid-form, it is now possible to relate structural and NMR parameters in a quantitative fashion. Several modelling and simulation techniques have been proposed to describe the influence of intermolecular interactions in the solid state on chemical shifts. In the cluster model, neighbouring molecules or fragments are considered explicitly during the chemical-shielding calculations. However, modelling a solid as a ‘large molecule’ or a cluster has many difficulties. The choice of the cluster, in particular its termination, is critical, as the calculations must be maintained at a manageable size. It is thus more efficient to exploit the translational repetition in crystals. In the last decade, the gauge-including projector-augmented wave (GIPAW) procedure has been developed for the prediction of the magnetic-resonance parameters in fully periodic solids.13 The wide applications of GIPAW-based calculations to “NMR crystallography” are now well documented.12,14 Note also that nucleic acid components have often been used as model compounds for the development and testing of computational methods. For example, the GIPAW approach has been tested against cluster calculations for solid isocytosine, where the cluster modelling was clearly found to be inferior.15
In this paper, we review recent applications (mostly after 2000) of solid-state NMR and NMR crystallography in studies of the structure of nucleic acid components, their intra- and intermolecular interactions, and dynamics.
Structure of nucleic acids components
One of the major conformational variables in nucleic acids is the pucker of the (deoxy)ribose ring. High-resolution X-ray studies have shown that the most common ring conformations in DNA are 3′-endo and 2′-endo. The pucker is inextricably linked to the helix geometry. For instance, the conformation is C2′-endo and C3′-endo in the B-form and the A-form of DNA respectively. The conformations of individual monomers in nucleic acids are thus important for their biological function.In the determination of nucleic acid structure by solution-state NMR, the backbone has a relatively low density of constraints because of the difficulty of obtaining conformational parameters from J couplings or NOE information within crowded spectral regions, particularly for large nucleic acids. For example, determining the backbone torsion angle γ(O5′–C5′–C4′–C3′) from 3JHH through the measurement of JH4′–H5′ and JH4′–H5′′ is often impractical because of the severe spectral overlapping of H5′ and H5′′ resonances, the difficulty in their stereo assignment, as well as poor detection because of the proximity of the water peak.16 Fig. 1 shows the conventional nucleotide atom numbering and torsion angle definitions.
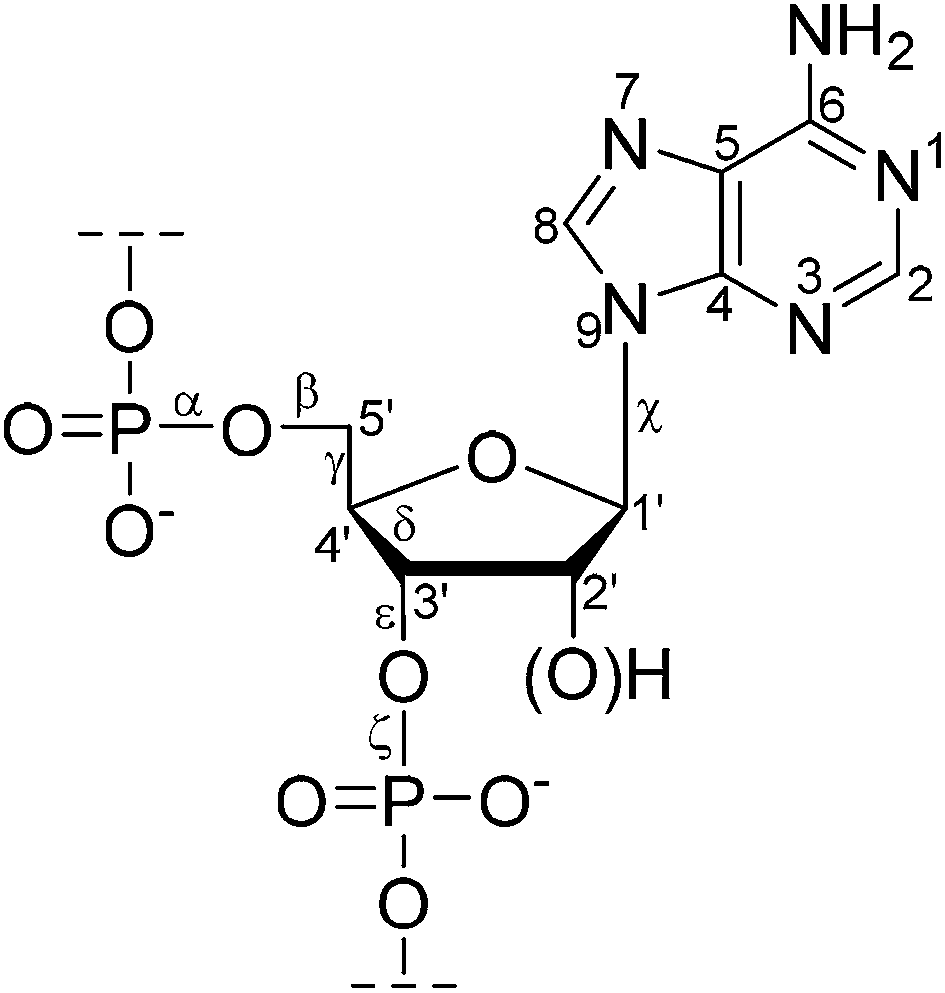 | ||
| Fig. 1 The structure of a (2′-deoxy)ribonucleotide fragment with atom numbering and selected torsion angles. | ||
In the solid, all of the major forms of DNA are accessible, either as fibres or as crystalline oligomers. In addition, crystalline nucleosides and nucleotides with a variety of ring puckers are known. Chemical shifts for a series of solid nucleosides and nucleotides with different deoxyribose ring conformations have been measured and the 13C chemical shifts were found to be related in a direct way to the ring pucker; 3′-endo conformers have significantly lower C3′ and C5′ chemical shifts (5–10 ppm) relative to comparable 3′-exo and 2′-endo conformers.4 The latter two conformers were distinguished by smaller, but still significant, differences in the carbon chemical shifts at the C2′ and C4′ positions. The same trends have also been observed for chemical shifts calculated by DFT methods for isolated nucleosides.17 CP-MAS NMR spectroscopy has also been used to investigate the dependence of 13C chemical shifts on specific conformational parameters of a variety of RNA nucleosides and nucleotides. It was shown that 13C chemical shifts can be used to determine sugar pucker and glycosidic (χ) and exocyclic (γ) angles in these systems with a high degree of certainty.18
Similarly, the dependence of 13C chemical shifts of the sugar ring on backbone torsion angle γ(O5′–C5′–C4′–C3′) and δ(C5′–C4′–C3′–O3′) as well as the sugar pucker, has been determined using crystalline nucleosides and nucleotides. The experimental data agreed well with DFT-calculated chemical shifts, implying that 13C chemical shifts are a useful tool for the determination of nucleic acid structure. The chemical shifts of C3′, C4′, and C5′ may be used for a reliable determination of the backbone torsion angles and the sugar pucker in most cases.16
An unusual DNA structure in Pf1 bacteriophage has been characterised by solid-state NMR. On the basis of experimental chemical shifts, obtained with dynamic-nuclear-polarisation-enhanced spectroscopy, it was concluded that Pf1 DNA exhibits a 2′-endo conformation because of its high C3′ and C5′ chemical shifts. The 13C and 15N chemical shifts of the DNA bases fall outside their typical regions in DNA, pointing to an absence of Watson–Crick hydrogen bonding.19 For example, adenine C4 and C5, and thymine C2 and C5 had unusually high chemical shifts, falling 1–2 ppm above the range of chemical shifts observed in B-DNA and cytosine C4, and guanosine C4 and C5 had unusually low chemical shifts. These observations were consistent with the absence of hydrogen bonding previously observed for thermal melting of DNA duplexes.20
The chemical shift of the 31P nucleus in the backbone of nucleic acids is influenced by the torsion angle ζ, which, at least in the B-type of nucleic acids, is either in the gauche region (approximately −60°, BI class) or in the trans region (approximately 180°, BII class). A phosphorus isotropic chemical shift difference of 1.8 ppm between the two classes has been extracted from 31P CP-MAS spectra of model solid oligonucleotides, the BII phosphorus atom having higher chemical shift than the BI.21 This study used macroscopically oriented samples, with fibres parallel to the rotor axis, allowing the orientation of the phosphate group with respect to the fibre axis to be determined. Although the BI ↔ BII conformational exchange is always fast in solution, a range of 31P shifts is observed in solution which is consistent with some systems existing predominantly in one conformation and others in a distribution, with the average 31P shift being determined by the BI/BII ratio.22,23
Chemical shift information can, however, be difficult to interpret due to non-local effects. Torsion angles may be more directly estimated by solid-state NMR using experiments that are sensitive to the relative orientations of nuclear spin interaction tensors. For example, experiments exploiting the evolution of a double quantum coherence under the heteronuclear local fields of neighbouring spins have been used to measure the δ torsion angles of two 2′-deoxynucleosides doubly 13C-labelled at the C3′ and C4′ positions.24 Similarly, the H1′–C1′–C6–H6 projection torsion angle defining the relative orientation of the nucleoside pyrimidine and ribose rings in uniformly labelled [13C,15N]uridine has been estimated by selective excitation of 13C double-quantum coherences under MAS at rotational resonance.25
Rotational echo double resonance (REDOR) is a solid-state NMR technique used to measure dipolar couplings and hence distances between pairs of different nuclear spins, which is frequently applied to biological structure problems. The distance range accessible by REDOR generally exceeds that of NOE or residual dipolar coupling measurements in solution. For example, the high magnetogyric ratio of 31P and 19F nuclei means that 31P,19F dipolar couplings are relatively strong, and 31P–19F distances of up to 16 Å have been measured.26 The high number of the phosphodiesters in the backbone of nucleic acids results in poorly resolved 31P NMR spectra. To enable site-specific detection of 31P–19F distances, a single phosphate group has been replaced by a phsphorothioate group, and fluorinated nucleotides have been placed in specific positions of model oligonucleotides.27 31P–19F REDOR has also been used to monitor changes in minor groove width of a DNA oligomer upon binding of the drug distamycin A at different stoichiometries (Fig. 2).28
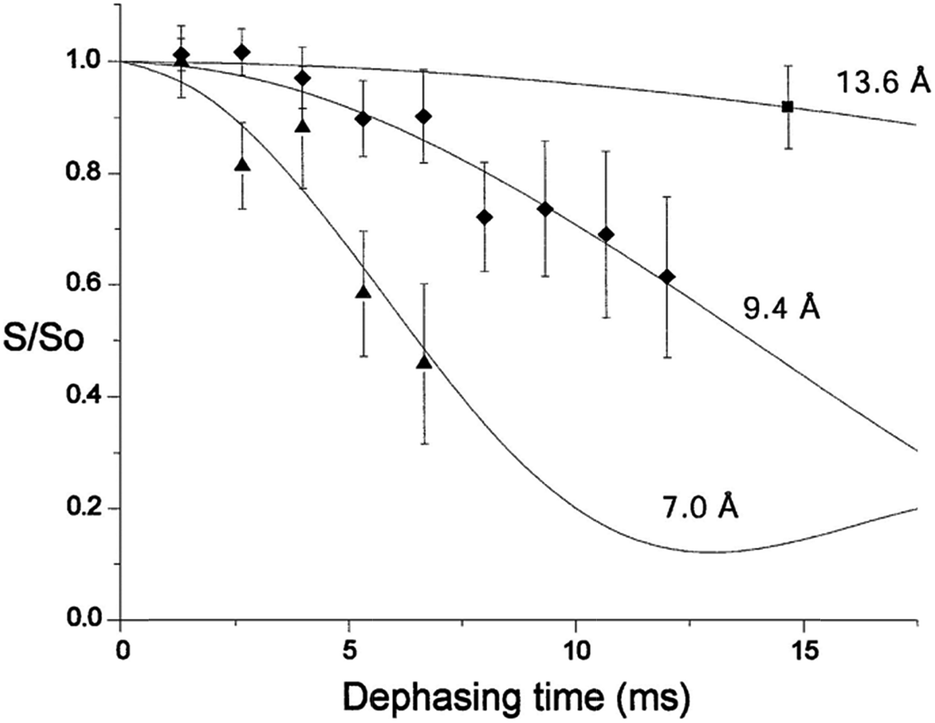 | ||
Fig. 2 31P–19F REDOR dephasing curves for a selectively fluoro- and phosphorothioate-substituted DNA and its 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 distamycin complexes. Solid lines represent expected decay curves based on simulations. Diamonds mark data for the unbound DNA, triangles for the 1:1 distamycin:DNA complex and the square for the 2:1 distamycin:DNA complex.28 Copyright Oxford University Press. Reproduced with permission. :1 and 2:1 distamycin complexes. Solid lines represent expected decay curves based on simulations. Diamonds mark data for the unbound DNA, triangles for the 1:1 distamycin:DNA complex and the square for the 2:1 distamycin:DNA complex.28 Copyright Oxford University Press. Reproduced with permission. | ||
Frequency-selective 31P–13C REDOR has been used to determine Pα–C8, Pβ–C8, and Pγ–C8 distances in ATP within the Na, K-ATPase enzyme. These distances were then used to propose the ATP conformation in the enzyme.29 These distances were compatible with a previous 13C-detected proton spin diffusion experiment, which was used to detect contacts between ATP and the binding site of the enzyme. The P–C distances followed the order C2 > C8 > ribose, which is consistent with the adenine ring of ATP being in contact with the binding site and the ribose ring being relatively exposed.30
Solid-phase synthesis has become the method of choice for producing oligonucleotides of defined sequence. 31P CP-MAS experiments have been used to monitor the solid-phase oligonucleotide-elongation reactions. The technique readily distinguishes the oxidation state of the phosphorus atom (phosphate/phosphite), the presence or absence of a protecting group attached to phosphorus, or phosphate vs. phosphorothioate groups.31 Similarly, 31P CP-MAS experiments have been used to study the complexation of 2′-deoxyadenosine-5′-phosphate (dAMP) with the surface of a mesoporous aluminium oxide. A single 31P resonance was observed upon complexation. However, 27Al MAS spectra show both tetrahedral and octahedral aluminium environments expected for the mesoporous alumina. 27Al–31P REDOR experiments revealed that the phosphate group of dAMP interacts predominantly with the octahedrally coordinated aluminium species at the surface. A comparison of experimental 31P CSA tensor parameters (obtained by the analysis of spinning side-bands) with those calculated for model clusters indicated that the binding was via a monodentate complex.32
Hydrogen bonding
The potential of nucleobases to form well-defined hydrogen-bonded base pairs is not only a major determinant of nucleic acid structure, but is also fundamental to important biological processes, such as replication and transcription. In addition, hydrogen-bond interactions between nucleobases and amino acid side chains are believed to play a crucial role in the recognition of specific nucleotide sequences by DNA-binding proteins.33,34 Detailed characterization of hydrogen bond interactions between biomolecular building blocks has, therefore, been the subject of numerous experimental and theoretical studies. NMR spectroscopy is one of the most powerful tools to study the strength and geometry of hydrogen bonds, although the study of hydrogen-bond interactions of small molecules by NMR is often hampered by the fast exchange of species in solution. Useful insight into the hydrogen bond strength and geometry is also obtained by comparing experimental NMR parameters with theoretical predictions.35,36Solid-state NMR has recently been used to identify ribbon-like and quartet-like self-assembly in guanosine derivatives by means of 1H chemical shifts and proton–proton proximities, as identified in 1H double-quantum/single-quantum correlation experiments (which used combined rotation and multiple-pulse spectroscopy (CRAMPS) to improve 1H spectral resolution). The NH proton chemical shift was observed to be higher (13–15 ppm) for ribbon-like self-assembly compared to 10–11 ppm for a quartet-like arrangement, corresponding to a change from NH⋯N to NH⋯O intermolecular hydrogen bonding.37 Hydrogen-bond networks in organosilicas based on adenine and thymine have also been studied by 1H solid-state NMR; spatial connectivities between protons were established using 1H–1H double quantum MAS experiments, allowing the geometries of hydrogen-bonded base pairs to be determined.38
Solid isocytosine provides an unusual opportunity to study two different tautomers of isocytosine, as they crystallize in a 1:1 ratio in a manner similar to that of the guanine and cytosine pairs in DNA. A combination of X-ray with solid-state NMR spectroscopic data and GIPAW calculations enabled precise structural parameters to be obtained, such as the geometries of intermolecular hydrogen bonds between isocytosine molecules, and by analogy Watson–Crick nucleic acid G–C base pairs. 1H chemical shifts of free NH and NH involved in the intermolecular hydrogen bond differ by 3 ppm (Fig. 3). In solution, the tautomers of isocytosine are in a fast equilibrium, and only averaged NMR parameters can be obtained.15
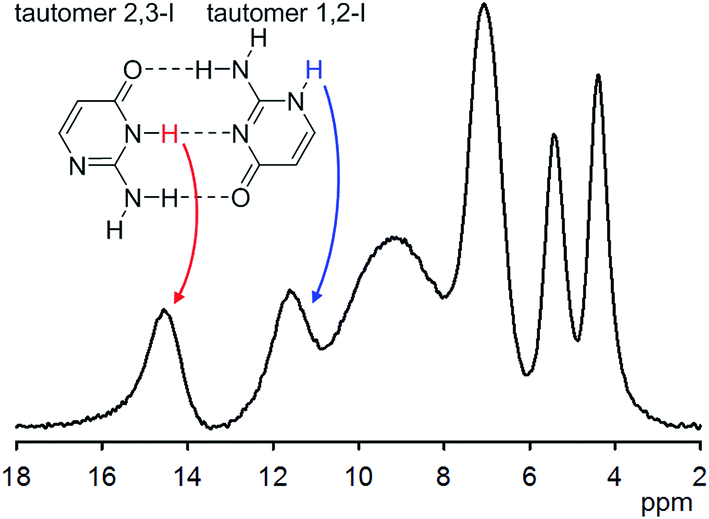 | ||
| Fig. 3 1H NMR spectrum of solid isocytosine acquired at 65 kHz MAS. Two tautomers in 1:1 ratio are present in the solid form. The chemical shift of the hydrogen-bound NH is substantially different from that of the free NH. | ||
Weak hydrogen bonding C–H⋯O in solid uracil has been investigated in a study that related experimentally determined 1H, 13C, and 15N chemical shifts with first-principles calculations. The effects of intermolecular interactions were quantified by comparing shifts calculated for isolated molecules, molecular planes, and a full crystal. Isolated molecule to plane changes in the 1H chemical shifts of 2 ppm were determined for the CH protons involved in the weak hydrogen bonding; this compares to changes of ca. 5 ppm for the NH protons involved in conventional NH⋯O hydrogen bonding.39 Similarly, the effects of conventional and weak hydrogen bonds on the principal components of 1H, 13C, and 15N chemical shift tensors and 14N and 17O electric field gradients of uracil atoms have been determined experimentally (for 13C and 15N) and computationally.40
The very low natural abundance of 17O limits oxygen NMR studies of nucleic acids components to 17O-enriched samples. For example, 17O NMR parameters have been determined for site-specifically 17O-enriched nucleic acid bases.41–43 The 17O NMR tensors were found to be highly sensitive to the nature of the intermolecular interactions in the solid state. The solid-state NMR determination of NMR interaction tensors of the carbonyl oxygen (O6) of guanine in two 17O-enriched guanosine derivatives has been reported. The 17O chemical-shift tensor and quadrupolar-coupling tensor were found to be very sensitive to the presence of hydrogen bonding and ion-carbonyl interactions, with the effect from ion–carbonyl interactions being several times stronger than that from hydrogen-bonding interactions.44
Although J couplings are not normally resolved in solid-state NMR spectra (because the observed linewidth is usually larger than the magnitude of the coupling), spin-echo based experiments often allow J couplings as small as 3.8 Hz to be measured.45,46 A powerful application of the spin-echo MAS technique is the quantification of hydrogen-bond mediated 2hJNN couplings, since it allows the identification of hydrogen-bonded partners, as well as the quantification of hydrogen-bond strengths and geometries.47 The detection of hydrogen bonds in the solid state via correlation peaks due to hydrogen-bond-mediated J coupling in a 15N refocused INADEQUATE spectrum has been reported for two guanosine derivatives. It was demonstrated that different N–H⋯N intermolecular hydrogen-bonding arrangements (quartet and ribbon) can be unambiguously identified in the spectra of the supramolecular guanosine structures.48 The intermolecular coupling constants in these structures have later been quantified by a 15N MAS spin-echo experiment.46 J-coupling-based experiments, such as INADEQUATE, provide direct information on bonding pathways (including through hydrogen bonds). However, experiments which use dipolar (i.e. through space) couplings, either directly between two 15N nuclei49 or indirectly via proton-driven spin diffusion,50 can also be used to identify inter-residue N–H⋯N hydrogen bonding e.g. in RNA.
Solid-state NMR can distinguish between polymorphs and is particularly suited for characterising subtle differences in crystal packing. For example, five polymorphs and one hydrate of 2-thiobarbituric acid have been characterised by 1D and 2D (1H, 13C, and 15N) solid-state NMR spectroscopy. The polymorphs differ in the tautomeric form of the compound; an enol form, a keto form, or a 1:1 mixture of both are present in the crystals. The tautomeric form is easily recognised by 13C CP-MAS spectroscopy, because the carbon chemical shift of C5 differs by ca. 40 ppm (see Fig. 4).51,52 Complete assignments of 1H and 13C resonances were obtained by combining 1D and 2D (homo- and heteronuclear data). 1H MAS NMR experiments provided information on hydrogen-bonded protons and their interaction strengths; the high 1H chemical shift values (close to 15 ppm) in two polymorphs suggested the presence of strong interactions, which is consistent with short hydrogen bonds observed by X-ray crystallography.51
 | ||
| Fig. 4 13C CP-MAS spectra of two polymorphs of 2-thiobarbituric acid. Polymorph III (top) contains keto form and polymorph II (bottom) enol form of 2-thiobarbituric acid. The asterisks indicate spinning side-bands. | ||
Co-crystals between a pharmaceutically active compound and a solid co-former are being widely investigated as an alternative to the use of drug salts for improving solid form properties (typically solubility). 13C NMR can straightforwardly verify the formation of a co-crystal, which will have an NMR spectrum which is distinct from that of the sum of the individual components. For example, 13C CP-MAS has been used to confirm co-crystal formation between acyclovir (an acyclic nucleoside antiviral drug) with both glutaric and fumaric acids. The carbon chemical shifts of acyclovir changed only slightly, which was rationalised in terms of the acyclovir molecule being involved in strong hydrogen bonding both in its pure and cocrystal forms. On the other hand, both glutaric and fumaric acid experience very different environments in the two cases, leading to changes in number of peaks due to changes of symmetry, and chemical shift changes of up to 5 ppm.53
Dynamics
Nucleic acids are highly flexible molecules that undergo functionally important structural transitions in response to external stimuli.54 Sequence-specific DNA flexibility plays essential roles in a variety of cellular processes that are crucial for gene packaging, expression and regulation.55,56 For example, intrinsic sequence-specific DNA flexibility is believed to play an important function in directing adaptive changes in DNA conformation that occur following protein and ligand recognition.57,58 It has also been proposed that a dynamic component or flexibility of a lesion nucleotide plays a significant role in the biomolecular recognition process of DNA lesions by repair enzymes.59,60 Similarly, many RNA functions are related to a multitude of functional dynamics.61The dynamics of nucleic acids span a broad range of time scales from picoseconds, where fast vibrational and librational motions occur, up to seconds, where catalytic function and global refolding take place. X-ray crystallography and solution NMR have contributed high-resolution structures of nucleic acids, but neither technique is suitable for an investigation of dynamics over such a broad range. On the other hand, solid-state NMR can probe motions with correlation times ranging over several orders of magnitudes.6,62 A particular advantage of working in the solid state is that it is unnecessary to deconvolute the effects of overall molecular motions. Pre-1991 solid-state NMR studies of DNA structure and dynamics have been reviewed by Alam and Drobny.9 This review also describes dynamic and motional processes in DNA and basic principles of NMR determination of DNA dynamics. Solid-state NMR may also help the interpretation of solution-state relaxation times by providing experimental chemical shift anisotropies. For example, the 31P chemical shift anisotropy of a 20mer RNA oligonucleotide under various salt and hydration conditions has been measured in order to interpret 31P relaxation data in solution.63 The principal components of 13C and 15N chemical-shift tensors in solid 3-, 7-, and 9-benzyladenine isomers have been determined and the influence of the substitution on the magnitude and orientation of the tensors has been discussed.64
Several isotopes can be utilised as probes for measuring dynamics in the solid state. One of the most useful isotopes is deuterium, because the solid-state NMR line shape and relaxation of deuterium spins are essentially dominated by a single mechanism – the interaction of the nuclear quadrupole moment with local electric field gradients. Using systematic isotopic labelling schemes, the local dynamics of the base, sugar, and backbone moieties of individual nucleotides within a sequence can be investigated with deuterium solid-state NMR experiments. A combination of deuterium line shape and relaxation data probes a wide range of motions from nanosecond time-scale dynamics (probed by relaxation measurements) to micro/millisecond time scales (from line shape measurements).65
For example, the internal motions of the nucleoside 2′-deoxythymidine in the solid state have been investigated by deuterium SS-NMR. The base position was found to be essentially rigid, even at elevated temperatures. On the other hand, T1 measurements on 2′,2′′-dideuterothymidine indicated the presence of two kinds of motion: (1) small-amplitude librations on the nanosecond time scale and (2) large amplitude jumping motions on the millisecond to microsecond time scale, which were hypothesised to be 2′-endo–3′-endo interconversion.66 Similarly two kinds of motion have been observed in a hydrated, non-crystalline sample of D-ribose selectively 2H-labelled at the 2′ position.67
Methylation of nucleotide bases is important for many biological processes. The HhaI system is a restriction-modification system consisting of a methyltransferase and endonuclease, which together act as a defense mechanism in prokaryotic systems, protecting the cell from invasive DNA. Deuterium SS-NMR has been used to understand and quantify the extent to which dynamics may assist proteins to recognise methylation sites distributed within DNA double helix, and to quantify the degree to which methylation perturbs the local dynamics of DNA (see Fig. 5 for an example of deuterium line shape analysis).65,68 The spectra obtained from DNAs selectively deuterated on the furanose ring within the GCGC moiety, recognised by the HhaI methyltransferase, indicated that all of these positions were structurally flexible. The furanose ring within the deoxycytidine that is the methylation target displayed the largest amplitude motion and ca. 10 times higher jump rates obtained by fitting the deuterium line shapes, whereas the furanose rings of nucleotides more remote from the methylation site had less mobile furanose rings. Furthermore, deuterium solid-state NMR revealed that methylation of the cytidine base reduces the amplitudes of motions of the phosphate – sugar backbone68,69 and changes the direction of the motions,70 even though the crystal structures displayed only small perturbations from unmethylated DNA. The deuterium solid-state NMR data were later compared with 13C solution relaxation measurements71 and with variable temperature solution 31P NMR72 and it was concluded that the local internal motions of the studied DNA oligomer in solution and in the hydrated solid state were virtually the same. On the basis of these results, it was hypothesized that local DNA dynamics promotes methylation by lowering energetic barriers for the conformational changes required for HhaI binding.
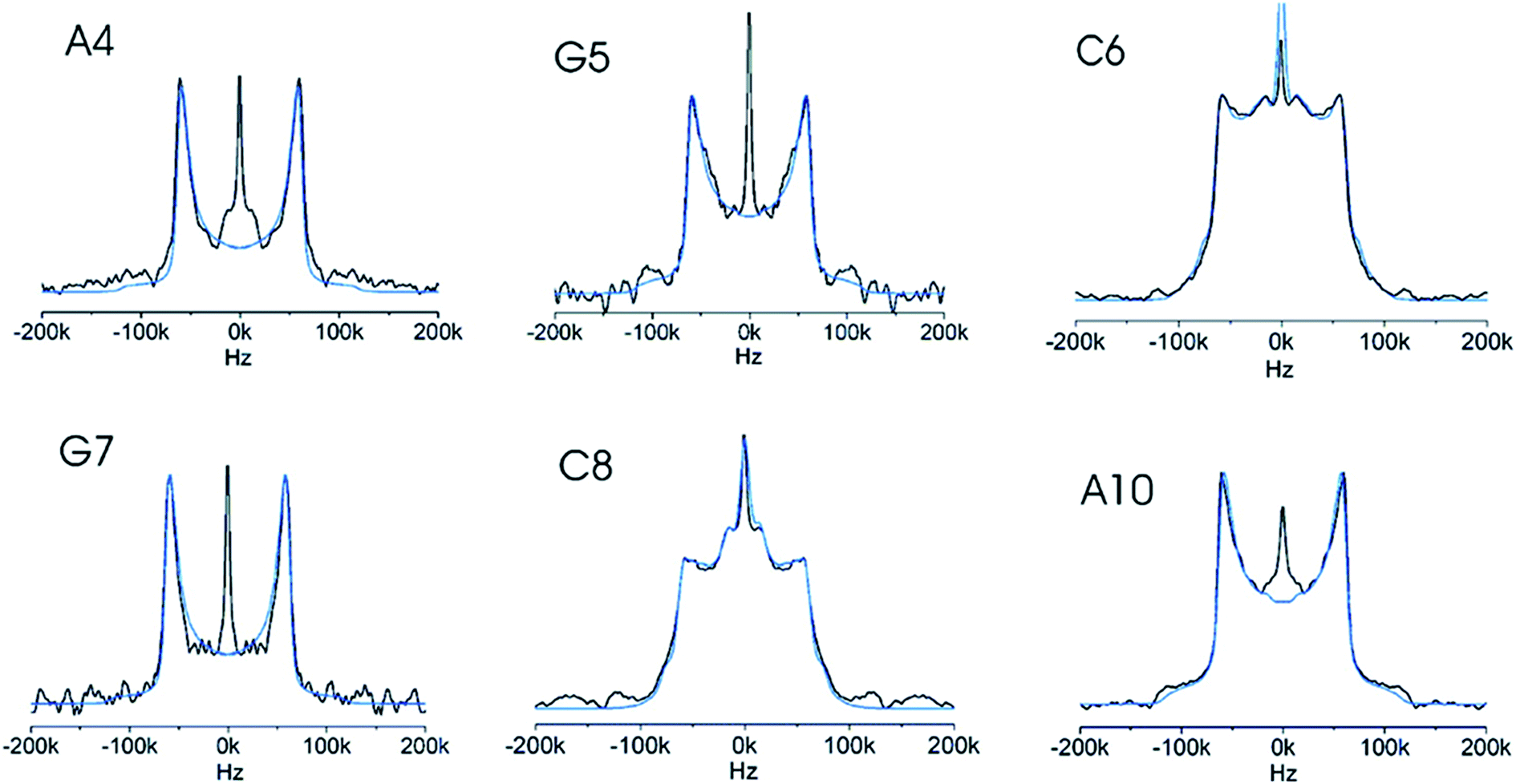 | ||
| Fig. 5 Six experimental deuterium line shapes (black) for individual selectively labelled sites in a DNA dodecamer with the simulation (blue) of each overlaid. The differences between the line shapes are caused by different local dynamics of the individual sites. Reprinted with permission from (ref. 65) Copyright 2008 American Chemical Society. | ||
In a similar study, solid-state 2H line shape and inversion recovery data were collected for six DNA samples containing deuterons at the H2′′ positions of various residues of a DNA dodecamer. The DNA was hydrated to 11–13 waters per nucleotide by vapour diffusion in a humidity chamber containing saturated salts in 2H-depleted water, to reach conditions where motions in the solid are very close to those observed in solution73 and to establish that the line shapes do not differ simply as a result of differential hydration. Remarkable variations in line shape and longitudinal relaxation times (T1Z) were observed between residues framing the methylation site and their neighbours. The residues close to the methylation site had shorter T1Z values of 20–30 ms and a noticeable modulation of the line shape, suggesting considerable motional averaging. Nearby residues were not nearly as flexible, as demonstrated by the line shapes and significantly larger T1Z (59–82 ms). It was established that all H2′′ nuclei experienced small amplitude librations (10°) of the C–D bond at frequencies faster than the quadrupolar interaction (174 kHz), and, in addition, H2′′ nuclei on the nucleotides close to the methylation site experienced large amplitude motions (36°) at similar frequencies. These effects are specific for the methylation target DNA sequence as other DNAs revealed no significant variation in T1Z or line shapes between individual residues.74
On the other hand, no significant differences in the local dynamics of the furanose ring within a uracil:adenine (U:A) base pair and a normal thymine:adenine (T:A) base pair have been revealed by deuterium solid-state NMR. The relaxation times were identical within the experimental error and the solid lineshapes were essentially indistinguishable. Therefore, U:A base pair furanose rings appeared to have identical dynamic properties as a normal T:A base pair, and the local dynamics of the furanose ring are unlikely to be the sole arbiter for uracil recognition and specificity in U:A base pairs.75
A solid-state deuterium NMR study of localised mobility at the C9pG10 step, the EcoRI restriction endonuclease target, in the DNA Dickerson dodecamer has been described both in crystalline and amorphous state.76,77 The furanose ring and helix backbone of dC9 display large amplitudes of motion on the 0.1 μs time scale, which contrasts with much smaller local dynamics in other nucleotides (dA5, dA6, dT7, and dT8) of the same dodecamer derived by earlier 2H NMR studies.78,79 The large amplitude motions occur only close to the site where the EcoRI restriction endonuclease binds and cleaves.
NMR interactions can be significantly influenced by fast molecular motions, such as vibrations. A theoretical study that combined DFT molecular dynamics simulations of a set of amino acids and nucleic acid bases with calculations of NMR parameters revealed the impact of vibrational motions on isotropic chemical shifts, chemical shift anisotropies (CSAs) and quadrupolar interactions. Re-orientation of the NMR tensors by molecular motion reduces the magnitudes of the NMR anisotropies, and inclusion of molecular dynamics significantly improved the agreement between calculated quadrupolar couplings and experimental values.80
NMR experiments together with molecular dynamics simulations and NMR calculations have been used to investigate mobility of water molecules and sodium ions in solid hydrates of two nucleotides. The structure of guanosine monophosphate system was found to be relatively rigid, with a well-ordered solvation shell of the nucleotide, while the water molecules in the uridine monophosphate system were shown to be remarkably mobile even at −80 °C. The disorder of water molecules was observed in the 13C, 31P, and 23Na solid-state NMR experiments as multiple signals for equivalent sites of the nucleotide corresponding to different local arrangements of the solvation shell. Deuterium NMR spectra of the samples recrystallized from D2O and molecular dynamics simulations also confirmed differences in water mobility between the two systems. The disordered solvation shell in UMP was proposed to be a good model for solvated nucleotides in general, with fast reorientation of water molecules and fluctuations in the hydrogen-bond network.81
Interactions with metal ions
Because (oligo)nucleotides are polyanions, their structure and biological function depends strongly on their association with metal ions. Metal ions are involved in almost every aspect of nucleic acid chemistry, ranging from neutralization of the anionic nucleic acids82 through specific stabilization of three-dimensional structures of nucleic acid molecules, up to their effect as cofactors in RNA-mediated catalysis.83 However, the dynamic non-covalent nature of these interactions has hampered the development of accurate and quantitative descriptions.84 Direct detection of light alkali metal ions by diffraction techniques is challenging, especially for sodium cations, because their X-ray scattering contributions are virtually identical to those of water, and Na+⋯O distances are only slightly shorter than strong hydrogen bonds between well-ordered water molecules.85 This often renders it impossible to identify Na+ ions, even with state-of-the-art diffraction techniques.Most of the metals that bind to nucleic acids are diamagnetic and possess significantly abundant isotopes that are NMR-active, making them potential targets for NMR. Unfortunately, the majority of these biologically significant isotopes also involve half-integer quadrupolar nuclei that provide limited information by solution-state NMR experiments due to the efficient quadrupolar relaxation that significantly broadens the NMR spectral lines. Moreover exchange of metals between bound sites and bulk solution is fast on the NMR timescale. These problems can be circumvented by carrying out the NMR experiments in the solid state, where the chemical exchange is stopped (or significantly reduced) and relaxation broadening quenched by the absence of rapid reorientations.86
Solid-state NMR has been frequently used for the characterisation of metal-ion interactions with nucleic acid components. For example, natural abundance 15N solid-state NMR spectra of complexes formed between Na+, Ba2+, and Cd2+ and guanosine-5′-monophosphate and inosine-5′-monophosphate demonstrated the great sensitivity of 15N shieldings to metal ion coordination. It was also shown that changes in the 15N chemical shift upon ion binding could be correlated with the strength and directionality of metal to nitrogen coordination.87
23Na NMR has been applied in several studies of the sodium salts of nucleotides. Usually, 1D 23Na MAS spectra do not exhibit resolved features from which information on the number of sodium sites and the associated NMR parameters can be readily extracted. In contrast, 2D 23Na multiple-quantum MAS (MQMAS) spectra usually display clearly distinct spectral regions corresponding to distinct sodium sites in the crystal lattice (see Fig. 6). From individual spectral cross-sections, it is possible to obtain three 23Na NMR parameters: the isotropic chemical shift δiso, quadrupolar coupling CQ and quadrupolar asymmetry ηQ. In some cases, the assignment of the NMR parameters to individual sites has been made on the basis of a simple correlation between CQ and the local ion-binding geometry.88,89 A partial assignment of the four non-equivalent sodium sites of Na2ATP was accomplished by incorporating 31P–23Na rotational echo double resonance (REDOR), variable temperature and relaxation methodologies onto the basic MQMAS high-resolution experiment.90 In the same paper, 23Na spin–lattice relaxation times were also determined and related to local mobility around the individual sodium sites. High-resolution 1D and 2D 23Na NMR spectra of deoxyguanosine-5′-monophosphate have also been obtained with the double-rotation (DOR) technique.91
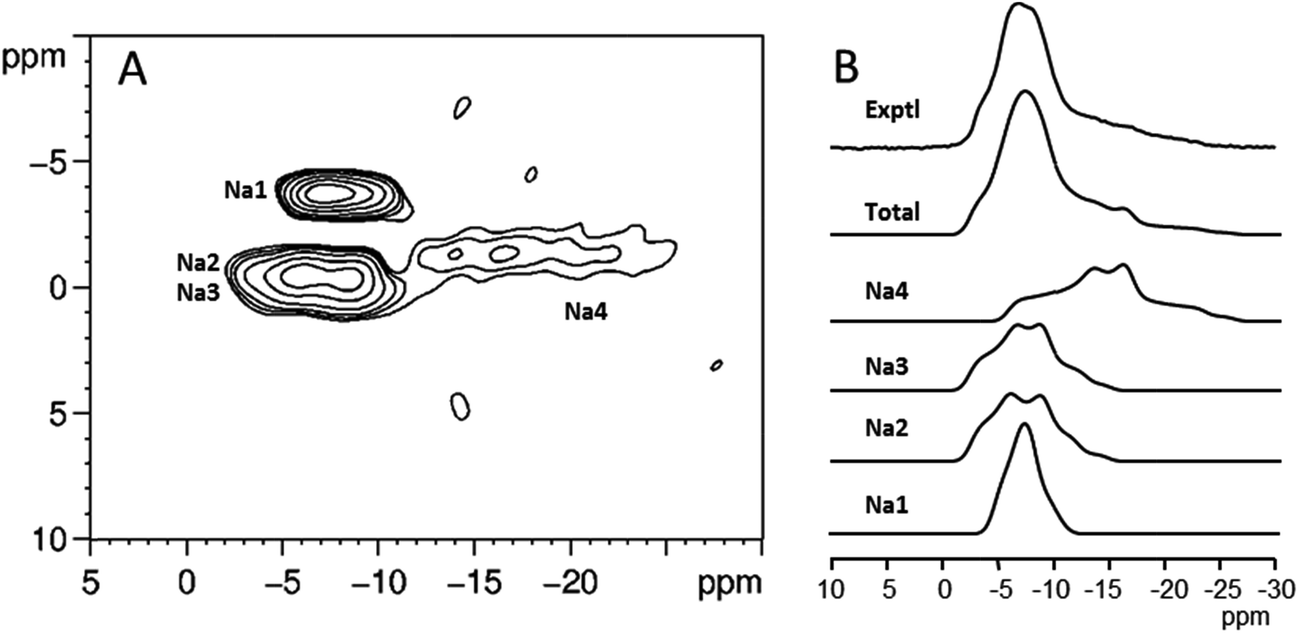 | ||
| Fig. 6 (A) 23Na MQMAS spectra of the disodium salt of guanosine-5′-monophosphate heptahydrate. (B) Individual cross-sections of the MQMAS spectrum, their sum, and the experimental 1D 23Na MAS spectrum. Figure adapted from results originally published in (ref. 89). | ||
A solid-state 23Na NMR study of monovalent cation (Li+, Na+, K+, Rb+, Cs+, and NH4+) binding to double-stranded calf thymus DNA at low relative humidity has been reported. Results from 23Na–31P REDOR experiment established that monovalent cations are directly bound to the phosphate groups of DNA and are partially dehydrated under these conditions. Quantitative thermodynamic parameters concerning the cation-binding affinity for the phosphate group were obtained by 23Na NMR titration experiments. These binding affinities were shown to be significantly different from those observed for the DNA in solution.92
Although magnesium is essential for the proper physiological folding of polynucleotides, direct NMR studies on this ion are complicated by its unfavourable nuclear properties (low natural abundance of 25Mg, low magnetogyric ratio, large quadrupolar moment). One possible magnesium analogue is [Co(NH3)6]3+, which binds to nucleic acids and is of similar size and shape as hexaaquamagnesium. The 59Co nuclide is a 100% naturally abundant isotope with relatively high magnetogyric ratio and moderate quadrupole moment. It has been demonstrated that 59Co MAS experiments on relatively small amount of tRNA can distinguish resonances corresponding to different metal binding environments. These characterisations were assisted by studies on model compounds and by 31P–59Co recoupling experiments.86
G-quadruplexes are DNA and RNA structural motifs composed of stacked G-quartets in which four guanine residues form a planar arrangement (Fig. 7). Because of their relevance to biological processes, such as DNA replication and transcription, these uncanonical structures are considered to be novel therapeutic targets and have also been identified as promising building blocks for DNA-based nanomaterials and nanodevices.93–95
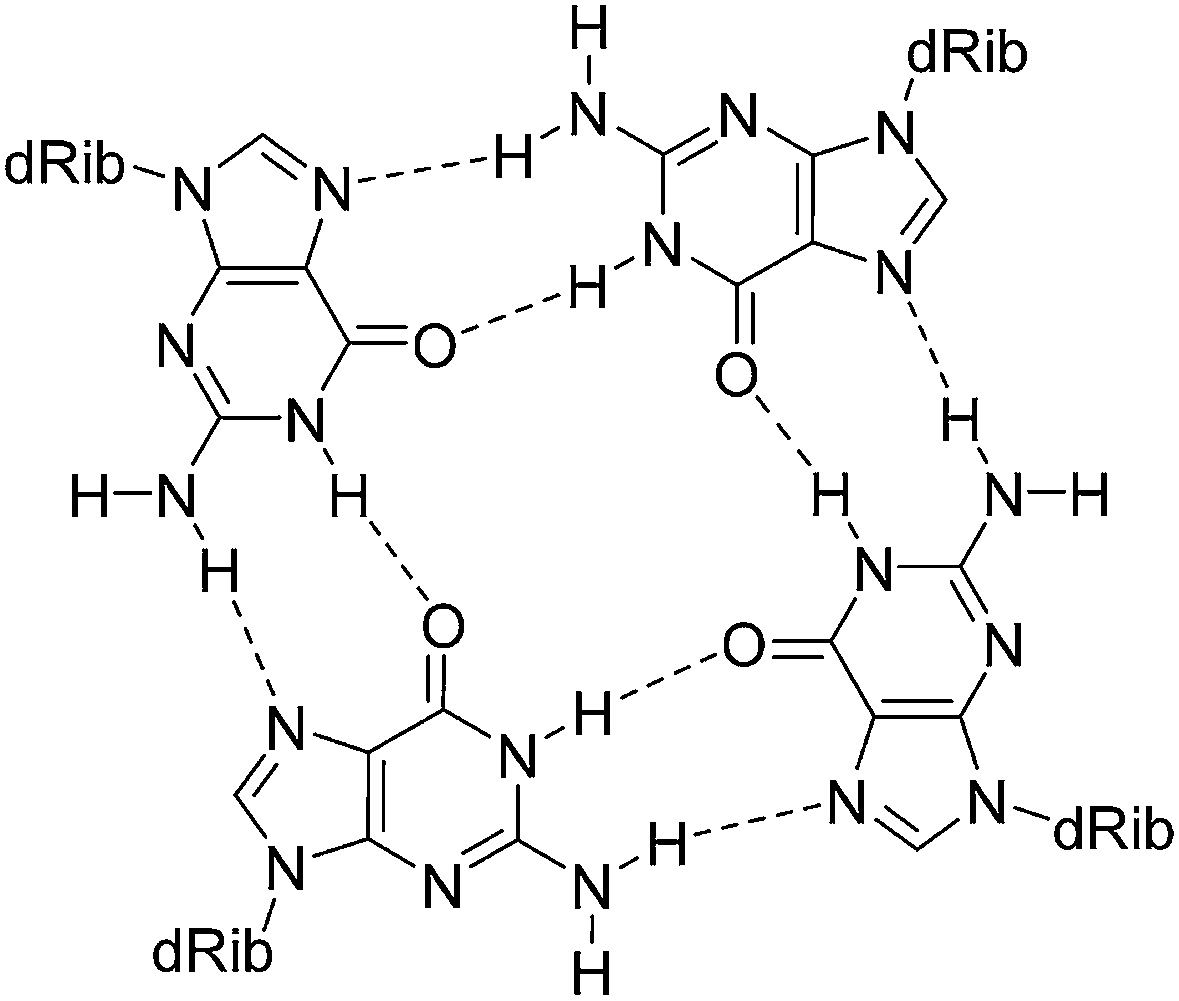 | ||
| Fig. 7 Diagram illustrating the cyclic hydrogen bonding in a G-quartet. The monovalent cation, which resides in or out of the guanine plane, is omitted. | ||
Alkali metal ions such as Na+ and K+ are known to play important roles in the formation, stability, and structural polymorphism of G-quadruplexes. Solution NMR has been used for studying alkali metal ion binding to G-quadruplexes. Although it is generally difficult to obtain site-specific information, with exceptions where spin-1/2 nuclear probes of 205Tl+ and 15NH4+ were used (see, for example, ref. 96–98), it has been recently shown that insight into the binding of sodium and potassium ions can also be obtained by NMR in solution.99 Solid-state NMR has emerged as a method for directly detecting alkali metal ions in these and related systems. Recently, solid-state NMR techniques have been successfully developed for the determination of cation coordination within G-quartet. Wu's group has studied the solid-state 23Na, 39K, and 87Rb NMR of guanosine complexes. For example, solid-state 23Na NMR has been used to determine the mode of Na+ binding to an Oxytricha nova telomeric DNA repeat. Using a 2D MQMAS 23Na experiment, three sodium cations residing inside the quadruplex channel were observed. Each of these sodium cations was sandwiched between two G-quartets.100 The utility of 2D MQMAS 23Na experiment in obtaining accurate site-specific information about ion binding in G-quadruplexes has also been confirmed in other studies.101–103
The relative affinity of monovalent cations for a stacking G-quartet structure was studied by solid-state NMR. Two major types of cations were found to be bound to the structure: one at the surface and the other within the channel cavity between two G-quartets. On the basis of solid-state 23Na results from a series of ion titration experiments, quantitative thermodynamic parameters concerning the relative cation binding affinity for each of the two major binding sites have been obtained.104 23Na NMR and quantum chemical calculations have also been used to determine the coordination of the sodium ion in a calix[4]arene–guanosine conjugate dimer, which was shown to form a single G-quartet at the centre of the structure with pentacoordinated sodium ion.105 23Na spin-echo experiments have been used to selectively suppress the phosphate-bound Na+ ions in a solid G-quadruplex, because they have shorter decoherence times than the G-quartet-bound sodium atoms.99
The presence of K+ ions in cells is believed to be crucial for the stability of telomeric G-quadruplex structures. The rather weak association between K+ ions and biological structures together with the low gyromagnetic ratio of 39K (spin 3/2) renders solution 39K NMR spectroscopy of limited utility. However, solid-state NMR detection of K+ ions bound to G-quadruplex structures has been shown to provide an unambiguous signature of potassium ions bound to G-quadruplex.106 It has been also proposed that 87Rb can be used as a surrogate of potassium for detecting K+ binding by solid-state NMR, because 87Rb has a much higher NMR sensitivity than 39K, but a similar radius.107
Conclusions
Despite their biological importance, many important issues related to the structure, dynamics and function of nucleic acids are not well understood. In this review, we have described recent applications of solid-state NMR and NMR crystallography to the study of nucleic acid components, focussing on applications where SS-NMR provides structural or dynamic information that is not accessible by other methods. Recent advances in experimental SS-NMR methods and DFT computations have opened new ways for studying nucleic acid systems. Limited motion in solids allows direct characterisation of individual conformations and intra- and intermolecular interactions. Furthermore, the local dynamics and interactions with the solvation shell and metal ions in solid hydrates are close to the hydration environment in solution, without the complication of overall molecular motion. Moreover, progress in the calculation of NMR parameters of solids enables NMR observables to be linked with structural models, greatly helping the interpretation of the experimental data. Solid-state NMR and NMR crystallography have thus become viable methods of determining the structure and local dynamics of nucleic acids and their components.Acknowledgements
This work was supported by the Czech Science Foundation (grant no. 13-24880S). The authors thank Prof. Gang Wu for providing the artwork for Fig. 6.References
- G. M. Blackburn, M. J. Gait, D. Loakes and D. M. Williams, Nucleic Acids in Chemistry and Biology: RSC, The Royal Society of Chemistry, Cambridge, 2006 Search PubMed.
- C. Simons, Q. P. Wu and T. T. Htar, Curr. Top. Med. Chem., 2005, 5, 1191–1203 CrossRef CAS PubMed.
- E. De Clercq and A. Holý, Nat. Rev. Drug Discovery, 2005, 4, 928–940 CrossRef CAS PubMed.
- R. A. Santos, P. Tang and G. S. Harbison, Biochemistry, 1989, 28, 9372–9378 CrossRef CAS PubMed.
- NMR Crystallography, ed. R. K. Harris, R. E. Wasylishen and M. J. Duer, John Wiley & Sons, Chichester, 2009 Search PubMed.
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid-State NMR: Basic Principles & Practice, Momentum Press, New York, 2012 Search PubMed.
- M. J. Duer, Introduction to Solid-State NMR Spectroscopy, Blackwell Publishing Ltd, Oxford, 2004 Search PubMed.
- A. Ramamoorthy, NMR Spectroscopy of Biological Solids, CRC Press, Boca Raton, 2005 Search PubMed.
- T. M. Alam and G. P. Drobny, Chem. Rev., 1991, 91, 1545–1590 CrossRef CAS.
- T. Gullion and J. Schaefer, Adv. Magn. Reson., 1989, 13, 57–83 Search PubMed.
- M. Kaupp, M. Bühl and V. G. Malkin, Calculation of NMR and EPR Parameters, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 6324, 245101 CrossRef.
- R. K. Harris, P. Hodgkinson, C. J. Pickard, J. R. Yates and V. Zorin, Magn. Reson. Chem., 2007, 45, S174–S186 CrossRef CAS PubMed.
- M. Dračínský, P. Jansa, K. Ahonen and M. Buděšínský, Eur. J. Org. Chem., 2011, 1544–1551 CrossRef.
- X. P. Xu, W. L. A. K. Chiu and S. C. F. Au-Yeung, J. Am. Chem. Soc., 1998, 120, 4230–4231 CrossRef CAS.
- J. M. Fonville, M. Swart, Z. Vokáčová, V. Sychrovský, J. E. Šponer, J. Šponer, C. W. Hilbers, F. M. Bickelhaupt and S. S. Wijmenga, Chem.–Eur. J., 2012, 18, 12372–12387 CrossRef CAS PubMed.
- M. Ebrahimi, P. Rossi, C. Rogers and G. S. Harbison, J. Magn. Reson., 2001, 150, 1–9 CrossRef CAS PubMed.
- I. V. Sergeyev, L. A. Day, A. Goldbourt and A. E. McDermott, J. Am. Chem. Soc., 2011, 133, 20208–20217 CrossRef CAS PubMed.
- S. R. LaPlante, E. A. Boudreau, N. Zanatta, G. C. Levy, P. N. Borer, J. Ashcroft and D. Cowburn, Biochemistry, 1988, 27, 7902–7909 CrossRef CAS PubMed.
- L. van Dam and M. H. Levitt, J. Mol. Biol., 2000, 304, 541–560 CrossRef CAS PubMed.
- B. Heddi, N. Foloppe, N. Bouchemal, E. Hantz and B. Hartmann, J. Am. Chem. Soc., 2006, 128, 9170–9177 CrossRef CAS PubMed.
- J. Přecechtělová, M. L. Munzarová, J. Vaara, J. Novotný, M. Dračínský and V. Sklenář, J. Chem. Theory Comput., 2013, 9, 1641–1656 CrossRef.
- L. van Dam, N. Ouwerkerk, A. Brinkmann, J. Raap and M. H. Levitt, Biophys. J., 2002, 83, 2835–2844 CrossRef CAS PubMed.
- S. G. Patching, R. Edwards and D. A. Middleton, J. Magn. Reson., 2009, 199, 242–246 CrossRef CAS PubMed.
- D. R. Studelska, C. A. Klug, D. D. Beusen, L. M. McDowell and J. Schaefer, J. Am. Chem. Soc., 1996, 118, 5476–5477 CrossRef CAS.
- E. A. Louie, P. Chirakul, V. Raghunathan, S. T. Sigurdsson and G. P. Drobny, J. Magn. Reson., 2006, 178, 11–24 CrossRef CAS PubMed.
- G. L. Olsen, E. A. Louie, G. P. Drobny and S. T. Sigurdsson, Nucleic Acids Res., 2003, 31, 5084–5089 CrossRef CAS PubMed.
- D. A. Middleton, E. Hughes and M. Esmann, Angew. Chem., Int. Ed., 2011, 50, 7041–7044 CrossRef CAS PubMed.
- D. A. Middleton, E. Hughes, N. U. Fedosova and M. Esmann, ChemBioChem, 2009, 10, 1789–1792 CrossRef CAS PubMed.
- P. M. Macdonald, M. J. Damha, K. Ganeshan, R. Braich and S. V. Zabarylo, Nucleic Acids Res., 1996, 24, 2868–2876 CrossRef CAS PubMed.
- R. A. Fry, K. D. Kwon, S. Komarneni, J. D. Kubicki and K. T. Mueller, Langmuir, 2006, 22, 9281–9286 CrossRef CAS PubMed.
- N. M. Luscombe, R. A. Laskowski and J. M. Thornton, Nucleic Acids Res., 2001, 29, 2860–2874 CrossRef CAS PubMed.
- E. M. B. Janke, H. H. Limbach and K. Weisz, J. Am. Chem. Soc., 2004, 126, 2135–2141 CrossRef CAS PubMed.
- M. Barfield, A. J. Dingley, J. Feigon and S. Grzesiek, J. Am. Chem. Soc., 2001, 123, 4014–4022 CrossRef CAS PubMed.
- E. Pauwels, D. Claeys, J. C. Martins, M. Waroquier, G. Bifulco, V. Van Speybroeck and A. Madder, RSC Adv., 2013, 3, 3925–3938 RSC.
- A. L. Webber, S. Masiero, S. Pieraccini, J. C. Burey, A. S. Tatton, D. Iuga, T. N. Pham, G. P. Spada and S. P. Brown, J. Am. Chem. Soc., 2011, 133, 19777–19795 CrossRef CAS PubMed.
- G. Arrachart, C. Carcel, J. J. E. Moreau, G. Hartmeyer, B. Alonso, D. Massiot, G. Creff, J. L. Bantignies, P. Dieudonne, M. W. C. Man, G. Althoff, F. Babonneau and C. Bonhomme, J. Mater. Chem., 2008, 18, 392–399 RSC.
- A. C. Uldry, J. M. Griffin, J. R. Yates, M. Perez-Torralba, M. D. S. Maria, A. L. Webber, M. L. L. Beaumont, A. Samoson, R. M. Claramunt, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 945–954 CrossRef CAS PubMed.
- S. K. Amini, H. Shaghaghi, A. D. Bain, A. Chabok and M. Tafazzoli, Solid State Nucl. Magn. Reson., 2010, 37, 13–20 CrossRef CAS PubMed.
- G. Wu, S. Dong, R. Ida and N. Reen, J. Am. Chem. Soc., 2002, 124, 1768–1777 CrossRef CAS PubMed.
- G. Wu, S. Dong and R. Ida, Chem. Commun., 2001, 891–892 RSC.
- G. Wu and S. Dong, J. Am. Chem. Soc., 2001, 123, 9119–9125 CrossRef CAS PubMed.
- I. C. M. Kwan, X. Mo and G. Wu, J. Am. Chem. Soc., 2007, 129, 2398–2407 CrossRef CAS PubMed.
- S. A. Joyce, J. R. Yates, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 12663–12670 CrossRef CAS PubMed.
- T. N. Pham, J. M. Griffin, S. Masiero, S. Lena, G. Gottarelli, P. Hodgkinson, C. Fillip and S. P. Brown, Phys. Chem. Chem. Phys., 2007, 9, 3416–3423 RSC.
- A. J. Dingley and S. Grzesiek, J. Am. Chem. Soc., 1998, 120, 8293–8297 CrossRef CAS.
- T. N. Pham, S. Masiero, G. Gottarello and S. P. Brown, J. Am. Chem. Soc., 2005, 127, 16018–16019 CrossRef CAS PubMed.
- J. Leppert, C. R. Urbinati, S. Hafner, O. Ohlenschlager, M. S. Swanson, M. Gorlach and R. Ramachandran, Nucleic Acids Res., 2004, 32, 1177–1183 CrossRef CAS PubMed.
- K. Riedel, J. Leppert, O. Ohlenschlager, M. Gorlach and R. Ramachandran, J. Biomol. NMR, 2005, 31, 331–336 CrossRef CAS PubMed.
- M. R. Chierotti, L. Ferrero, N. Garino, R. Gobetto, L. Pellegrino, D. Braga, F. Grepioni and L. Maini, Chem.–Eur. J., 2010, 16, 4347–4358 CrossRef CAS PubMed.
- M. Dračínský, E. Procházková, J. Kessler, J. Šebestík, P. Matějka and P. Bouř, J. Phys. Chem. B, 2013, 117, 7297–7307 CrossRef PubMed.
- G. Bruni, M. Maietta, L. Maggi, P. Mustarelli, C. Ferrara, V. Berbenni, C. Milanese, A. Girella and A. Marini, J. Pharm. Sci., 2013, 102, 4079–4086 CrossRef CAS PubMed.
- E. N. Nikolova and H. M. Al-Hashimi, J. Biomol. NMR, 2009, 45, 9–16 CrossRef CAS PubMed.
- H. G. Garcia, P. Grayson, L. Han, M. Inamdar, J. Kondev, P. C. Nelson, R. Phillips, J. Widom and P. A. Wiggins, Biopolymers, 2007, 85, 115–130 CrossRef CAS PubMed.
- F. Gimenes, K. I. Takeda, A. Fiorini, F. S. Gouveia and M. A. Fernandez, Genet. Mol. Res., 2008, 7, 549–558 CrossRef CAS PubMed.
- Z. Shajani and G. Varani, Biochemistry, 2008, 47, 7617–7625 CrossRef CAS PubMed.
- S. Fujii, H. Kono, S. Takenaka, N. Go and A. Sarai, Nucleic Acids Res., 2007, 35, 6063–6074 CrossRef CAS PubMed.
- J. T. Stivers, Prog. Nucleic Acid Res. Mol. Biol., 2004, 77, 37–65 CAS.
- W. Yang, Cell Res., 2008, 18, 184–197 CrossRef CAS PubMed.
- H. M. Al-Hashimi and N. G. Walter, Curr. Opin. Struct. Biol., 2008, 18, 321–329 CrossRef CAS PubMed.
- R. Tycko, Nuclear Magnetic Resonance Probes of Molecular Dynamics, Kluwer Academic Publishers, Dordecht, 1994 Search PubMed.
- J. Rinnenthal, C. Richter, S. Nozinovic, B. Furtig, J. J. Lopez, C. Glaubitz and H. Schwalbe, J. Biomol. NMR, 2009, 45, 143–155 CrossRef CAS PubMed.
- K. Maliňáková, L. Novosadová, M. Pipíška and R. Marek, ChemPhysChem, 2011, 12, 379–388 CrossRef PubMed.
- G. A. Meints, P. A. Miller, K. Pederson, Z. Shajani and G. Drobny, J. Am. Chem. Soc., 2008, 130, 7305–7314 CrossRef CAS PubMed.
- Y. Hiyama, S. Roy, J. S. Cohen and D. A. Torchia, J. Am. Chem. Soc., 1989, 111, 8609–8613 CrossRef CAS.
- A. C. LiWang, D. E. McCready, G. P. Drobny, B. R. Reid and M. A. Kennedy, J. Biomol. NMR, 2003, 26, 249–257 CrossRef CAS PubMed.
- G. A. Meints and G. P. Drobny, Biochemistry, 2001, 40, 12436–12443 CrossRef CAS PubMed.
- K. B. Geahigan, G. A. Meints, M. E. Hatcher, J. Orban and G. P. Drobny, Biochemistry, 2000, 39, 4939–4946 CrossRef CAS PubMed.
- K. Pederson, G. A. Meints, Z. Shajani, P. A. Miller and G. P. Drobny, J. Am. Chem. Soc., 2008, 130, 9072–9079 CrossRef CAS PubMed.
- D. Echodu, G. Goobes, Z. Shajani, K. Pederson, G. Meints, G. Varani and G. Drobny, J. Phys. Chem. B, 2008, 112, 13934–13944 CrossRef CAS PubMed.
- Y. Tian, M. Kayatta, K. Shultis, A. Gonzalez, L. J. Mueller and M. E. Hatcher, J. Phys. Chem. B, 2009, 113, 2596–2603 CrossRef CAS PubMed.
- J. M. Schurr, B. S. Fujimoto, R. Diaz and B. H. Robinson, J. Magn. Reson., 1999, 140, 404–431 CrossRef CAS PubMed.
- P. A. Miller, Z. Shajani, G. A. Meints, D. Caplow, G. Goobes, G. Varani and G. P. Drobny, J. Am. Chem. Soc., 2006, 128, 15970–15971 CrossRef CAS PubMed.
- M. N. Kinde-Carson, C. Ferguson, N. A. Oyler, G. S. Harbison and G. A. Meints, J. Phys. Chem. B, 2010, 114, 3285–3293 CrossRef CAS PubMed.
- M. E. Hatcher, I. LeTrong, R. Stenkamp and G. P. Drobny, J. Am. Chem. Soc., 2001, 123, 8874–8875 CrossRef CAS PubMed.
- M. E. Hatcher, D. L. Mattiello, G. A. Meints, J. Orban and G. P. Drobny, J. Am. Chem. Soc., 1998, 120, 9850–9862 CrossRef CAS.
- W. C. Huang, J. Orban, A. Kintanar, B. R. Reid and G. P. Drobny, J. Am. Chem. Soc., 1990, 112, 9059–9068 CrossRef CAS.
- T. M. Alam, J. Orban and G. P. Drobny, Biochemistry, 1991, 30, 9229–9237 CrossRef CAS PubMed.
- M. Dračínský and P. Hodgkinson, CrystEngComm, 2013, 15, 8705–8712 RSC.
- M. Dračínský, M. Šála and P. Hodgkinson, CrystEngComm, 2014, 16, 6756–6764 RSC.
- J. J. Howard, G. C. Lynch and B. M. Pettitt, J. Phys. Chem. B, 2011, 115, 547–556 CrossRef CAS PubMed.
- J. Müller, Metallomics, 2010, 2, 318–327 RSC.
- Nucleic Acid-Metal Ion Interactions, ed. N. V. Hud, Royal Society of Chemistry, Cambridge, 2009 Search PubMed.
- V. Tereshko, C. J. Wilds, G. Minasov, T. P. Prakash, M. A. Maier, A. Howard, Z. Wawrzak, M. Manoharan and M. Egli, Nucleic Acids Res., 2001, 29, 1208–1215 CrossRef CAS PubMed.
- C. V. Grant, V. Frydman, J. S. Harwood and L. Frydman, J. Am. Chem. Soc., 2002, 124, 4458–4462 CrossRef CAS PubMed.
- K. Barbarossou, A. E. Aliev, I. P. Gerothanassis, J. Anastassopoulou and T. Theophanides, Inorg. Chem., 2001, 40, 3626–3628 CrossRef CAS PubMed.
- G. Wu and A. Wong, in NMR Spectroscopy of Biological Solids, ed. A. Ramamoorthy, CRC Press, Boca Raton, 2006 Search PubMed.
- G. Wu and J. F. Zhu, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 61, 1–70 CrossRef CAS PubMed.
- C. V. Grant, D. McElheny, V. Frydman and L. Frydman, Magn. Reson. Chem., 2006, 44, 366–374 CrossRef CAS PubMed.
- F. A. Perras, I. Korobkov and D. L. Bryce, Phys. Chem. Chem. Phys., 2012, 14, 4677–4681 RSC.
- A. Wong, Z. M. Yan, Y. N. Huang and G. Wu, Magn. Reson. Chem., 2008, 46, 308–315 CrossRef CAS PubMed.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS PubMed.
- D. S. Liu, E. J. Cheng and Z. Q. Yang, NPG Asia Mater., 2011, 3, 109–114 CrossRef.
- R. Rodriguez, K. M. Miller, J. V. Forment, C. R. Bradshaw, M. Nikan, S. Britton, T. Oelschlaegel, B. Xhemalce, S. Balasubramanian and S. P. Jackson, Nat. Chem. Biol., 2012, 8, 301–310 CrossRef CAS PubMed.
- M. Trajkovski and J. Plavec, J. Phys. Chem. C, 2012, 116, 23821–23825 CAS.
- M. L. Gill, S. A. Strobel and J. P. Loria, J. Am. Chem. Soc., 2005, 127, 16723–16732 CrossRef CAS PubMed.
- P. Sket, A. Virgilio, V. Esposito, A. Galeone and J. Plavec, Nucleic Acids Res., 2012, 40, 11047–11057 CrossRef CAS PubMed.
- R. Ida and G. Wu, J. Am. Chem. Soc., 2008, 130, 3590–3602 CrossRef CAS PubMed.
- G. Wu and A. Wong, Biochem. Biophys. Res. Commun., 2004, 323, 1139–1144 CrossRef CAS PubMed.
- A. Wong, J. C. Fettinger, S. L. Forman, J. T. Davis and G. Wu, J. Am. Chem. Soc., 2002, 124, 742–743 CrossRef CAS PubMed.
- G. Wu and A. Wong, Chem. Commun., 2001, 2658–2659 RSC.
- D. Rovnyak, M. Baldus, G. Wu, N. V. Hud, J. Feigon and R. G. Griffin, J. Am. Chem. Soc., 2000, 122, 11423–11429 CrossRef CAS.
- A. Wong and G. Wu, J. Am. Chem. Soc., 2003, 125, 13895–13905 CrossRef CAS PubMed.
- A. Wong, F. W. Kotch, I. C. M. Kwan, J. T. Davis and G. Wu, Chem. Commun., 2009, 2154–2156 RSC.
- G. Wu, A. Wong, Z. H. Gan and J. T. Davis, J. Am. Chem. Soc., 2003, 125, 7182–7183 CrossRef CAS PubMed.
- R. Ida and G. Wu, Chem. Commun., 2005, 4294–4296 RSC.
| This journal is © The Royal Society of Chemistry 2015 |