
Hafnium catalysts for direct alkene epoxidation using molecular oxygen as oxidant†
Abstract
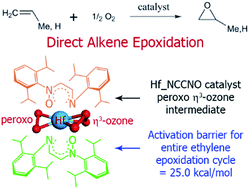
New zirconium (Zr) based organometallic catalysts for direct olefin epoxidation using O2 as oxidant without coreductant were introduced in a previous computational study (T. A. Manz and B. Yang, RSC Adv., 2014, 4, 27755–27774). In this paper, we use density functional theory (DFT) to study three Hf-based catalysts with the same bis(bidentate) ligands as the preceding Zr-based catalysts: (a) the diimine ligand N(Ar)–CH–CH–N(Ar) aka NCCN, (b) the imine–nitrone ligand N(Ar)–CH–CH–N(Ar)–O aka NCCNO, and (c) the dinitrone ligand O–N(Ar)–CH–CH–N(Ar)–O aka ONCCNO [Ar = –C6H3–2,6-iPr2]. Complete reaction cycles and energetic spans (i.e., effective activation energies for the entire catalytic cycle) are computed for propene epoxidation. For Hf_NCCNO and Hf_ONCCNO, the reaction cycles are similar to the Zr-based analogs and the formation of η3-ozone intermediates is still crucial. Hf_ONCCNO has a large enthalpic energetic span (60.4 kcal mol−1) due to forming inert octahedral complexes as the catalyst resting state. Our calculations predict an energetic span ≥40 kcal mol−1 for Hf_NCCN, which indicates it will not be a good catalyst. Computed enthalpic energetic spans of ∼30 kcal mol−1 are achieved for the Hf_NCCNO and Zr_NCCNO catalysts; however, transfer of allylic hydrogen from the reaction product forms a low energy deactivation product. Therefore, the Hf_NCCNO and Zr_NCCNO catalysts are only suitable for direct epoxidation of alkenes that do not have any allylic hydrogen atoms. As an example of an alkene with no allylic hydrogens, we computed enthalpic energetic spans (kcal mol−1) for direct ethylene epoxidation of (a) 25.0 for Hf_NCCNO, (b) 30.2 for Zr_NCCNO, and (c) 52.7 for Zr_NCCN.
 Please wait while we load your content...
Please wait while we load your content...

