
Large mesoporous carbons decorated with silver and gold nanoparticles by a self-assembly method: enhanced electrocatalytic activity for H2O2 electroreduction and sodium nitrite electrooxidation†
X. J. Yangab,
Y. H. Wangab,
J. Baib,
X. Y. He*a and
X. E. Jiang*b
aChina West Normal University, Nanchong 637002, China. E-mail: hexysctc@yahoo.com.cn; Fax: +86 431 85262378; Tel: +86 431 85685653
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Science, Changchun 130022, China. E-mail: jiangxiue@ciac.ac.cn; Fax: +86 431 85262378; Tel: +86 431 85685653
First published on 2nd December 2014
Abstract
We report here the synthesis of silver and gold nanoparticles onto poly (diallyldimethyl ammoniumchloride, PDDA)-functionalized large mesoporous carbon (LMC) by a simple self-assembly method. Characterization of these nanocomposites by transmission electronic microscopy and electrochemical techniques is described. The gold nanoparticle-decorated PDDA-functionalized LMC (AuNPs/PDDA–LMC) showed an enhanced electrocatalytic response toward sodium nitrite oxidation with a wider linear range and higher sensitivity that is even comparable with Pt-based sensors. The silver nanoparticle-decorated PDDA-functionalized LMC (AgNPs/PDDA–LMC) exhibited a remarkable catalytic performance toward H2O2 reduction. At an applied potential of −0.4 V, the designed sensor had a low detection limit of 6.5 μM and wide linear response range from 20 μM to 9.62 mM with a fast response time of less than 2 s. The easy fabrication and excellent electrocatalytic capability of the LMC-based metal nanoparticle hybrids can be expected to be applied in the detection of other compounds.
Introduction
In recent years, considerable attention has been paid to mesoporous carbon materials due to their combination of high specific surface areas, large pore volume, chemical inertness, and good mechanical and thermal stabilities.1–4 These properties endow mesoporous carbon materials with promising performances for potential applications in the fields of electrocatalysis, adsorption, energy storage and electrochemical double capacitors.5–10 Owing to the large surface area, mesoporous carbon materials have been an attractive choice as the substrates to enhance the functionality of materials.11–13 For instance, the complex of noble metal nanoparticles with mesoporous carbon materials potentially paves a new way to enhance their electronic, chemical, and electrochemical properties.14–16 In comparison with mesoporous carbon materials, one easily-prepared large mesoporous carbon (LMC) has also shown superior electrocatalytic performance than traditional carbon materials.17–19 Therefore, the new developed LMC with large surface area, pore size and low cost may be a good candidate for host–guest chemistry to support metal nanoparticles for electrocatalysts.The applications of nanosized noble metals have gained considerable attention in the past few years due to their size-dependent electrical, chemical and optical properties.20,21 However, metal nanoparticles usually suffer from aggregation issues during their synthesis and storage processes, which often prohibits the application of the particles. Therefore, to obtain high dispersibility of the nanoparticles, many successful methods have been used to stabilize the nanoparticles, such as capping by polyelectrolytes,22 and stabilized in carbon- or silicate-based matrix.23,24 But, to the best of our knowledge, only one example for LMC-based metal nanoparticles hybrid has been reported till now. Moreover, the distribution of metal nanoparticles on the supporting substrate is quite poor based on chemical reduction method. Therefore, it is still a challenge to synthesize LMC-based metal nanoparticles hybrids with high loading and uniform distribution.
Molecular self-assembly has become an attractive method in fabrication of nanodevices due to its simplicity and controllability. By virtue of the interactions between the atoms, molecules, polymers, or particles of the building blocks, it is easy to get large-area nanomaterials with unique properties.25 In this work, we demonstrated the possibility to construct the LMC-based metal nanoparticles with high loading and uniform distribution by self-assembled method. Poly-diallyldimethyl ammoniumchloride (PDDA), a water-soluble cationic polyelectrolyte with adhesion and film-forming properties, was used as a dispersant to functionalize the LMC since positively charged PDDA can be easily coated onto negatively charged LMC through electrostatic interactions. Typical citrate-capped Ag and Au nanoparticles were then chosen as model nanomaterials to interact with the positively charged PDDA-functionalized LMC. The adhesion and film-forming properties of PDDA play an essential role in the construction of stable and useful bio-interface. This also provides a convenient self-assembly method for the hybridization of LMC.
To demonstrate the electrochemical-enhanced property of the resulted nanocomposites, the high density Ag and Au-modified LMC were used to construct hydrogen peroxide (H2O2) and sodium nitrite sensor, respectively. Hydrogen peroxide is a residual agent in food disinfection, a waste product of industry and a valuable marker of many oxidative biological reactions.26–29 Sodium nitrite is classified as an environmentally hazardous species and always appears in soils, waters, and foods.30,31 Therefore, detection of hydrogen peroxide and sodium nitrite is of great importance. Combining the advantages of metal nanoparticles and PDDA-coated LMC, the Ag/LMC-based sensor showed excellent electrocatalytic property to H2O2 with a fast amperometric response time, wide linear ranges and low detection limit, and the Au/LMC-based senor showed excellent catalytic activity toward the oxidation of sodium nitrite.
Experimental section
Materials and instruments
NaH2PO4, Na2HPO4, glucose, citric acid, sucrose, NaCl, HCl, H2O2 (30 wt%), AgNO3 and SiO2 were purchased from Beijing Chemical Corp. (Beijing, China). Chitosan, Nafion® (5 wt%), ascorbic acid (AA), dopamine (DA), acetaminophen (AP) and uric acid (UA) were obtained from Aladin Ltd (Shanghai, China). Poly (diallyldimethyl ammoniumchloride) (PDDA) was from Shanghai Chemical Reagent Corp. (Shanghai, China). All chemicals were used without further purification. The water used throughout all experiments was purified through a Millipore system. Phosphate buffer saline (PBS) was prepared by mixing stock solutions of NaH2PO4 and Na2HPO4. A fresh solution of H2O2 was prepared daily.The transmission electron microscopy (TEM) measurements of the pre-prepared sample by dropping the dispersion on carbon-coated copper grid were made on a HITACHI H-8100 EM (Hitachi, Tokyo, Japan). The X-ray diffraction (XRD) pattern was collected on a Japan Rigaku D/Max-RA X-ray diffractometer. The sample for XRD characterization was prepared by placing the dispersion on a glass slide. Zeta potential measurements were performed using a Zetasizer Nano ZS (Malvern Instruments). Electrochemical experiments were performed with a CHI 830 electrochemical analyzer (CH Instruments, Chenhua Co., Shanghai, China). A three-electrode configuration was employed, consisting of a GC electrode (3 mm diameter) or a modified GC electrode serving as a working electrode, an Ag/AgCl (in saturated KCl solution) and a platinum wire served as the reference and counter electrodes, respectively.
Synthesis of PDDA-functionalized LMC
The LMC was synthesized based on the previous report.32 Then, the LMC was carboxylated by treatment with nitric acid for 3 h at 84 °C. After that, samples were centrifuged and washed with distilled water, then further dried at 40 °C overnight.The carboxylated LMC was functionalized with PDDA by the following process. 10 mg of carboxylated LMC was dispersed in 3 mL of 0.5 wt% PDDA aqueous solution containing 0.5 wt% NaCl and sonicated for 2 h, then stirred for 24 h, followed by washing and centrifugation. At last, the obtained PDDA/LMC was dispersed in 0.5 mL water.
Synthesis of citrate-capped Ag and Au nanoparticles
AgNPs were synthesized according to reported methods,33,34 in a typical preparation process, 0.53 mL of 0.1 M AgNO3 aqueous solution was added to 50 mL of water and heated rapidly to boiling. After that 1 mL of a 1% sodium citrate dihydrate was added under vigorous stirring for the preparation 20 nm AgNPs, and the resulting solution was held at boiling for 45 min.AuNPs were synthesized by the following steps:35 100 mL of HAuCl4 (1 mM) aqueous solution was refluxed under stirring and then 10 mL of trisodium citrate solution (38.8 mM) was quickly added in order to prepare 16 nm Au NPs. Then the solution was refluxed for additional 15 min. The resulting solution was fixed to 1 mM by evaporating excess water.
Fabrication of high density Ag and Au-modified LMC
40 μL of 1 mg mL−1 of PDDA–LMC was added to 4 mL of as prepared AgNPs or AuNPs under stirring. Then the solution was sonicated for 3 min before keeping overnight. The precipitate was washed several times and dispersed in 1 mL of water.Preparation of the modified electrodes
Glassy carbon electrode (GCE, 3 mm in diameter) was polished with 1.0 and 0.3 μm alumina slurry sequentially and then washed ultrasonically in water and ethanol for a few minutes, respectively. The cleaned GCE was dried in a stream of high-purity nitrogen. After that, 5.0 μL of AgNPs/PDDA–LMC or AuNPs/PDDA–LMC solution was dropped onto the surface of pretreated GCE and left to dry at room temperature to get nanocomposites-modified GCE, referred as AgNPs/PDDA–LMC/GCE or AuNPs/PDDA–LMC/GCE, respectively. Thereafter, 2 μL of chitosan (0.2%) was dropped on the surface of the as-prepared nanocomposites-modified GCE and allowed to dry at ambient conditions. As control experiments, PDDA–LMC, AgNPs, and AuNPs-modified GCEs were prepared with identical conditions.Results and discussion
Characterization of the prepared AgNPs/PDDA–LMC and AuNPs/PDDA–LMC
The procedure of the self-assembly of PDDA–LMC and citrate-capped AgNPs or AuNPs is schematically presented in Scheme 1. As described above, fist the prepared LMC were carboxylated by acid treatment. Then, PDDA–LMC conjugates were prepared by dispersing the carboxylated LMC in a positively charged PDDA aqueous solution for wrapping of LMC surface with positive charges. Finally, the cationic PDDA–LMC dispersion was directly mixed with the as-prepared citrate-capped AgNPs or AuNPs. AgNPs or AuNPs were quickly adhered to the surface of PDDA–LMC because of the electrostatic interaction.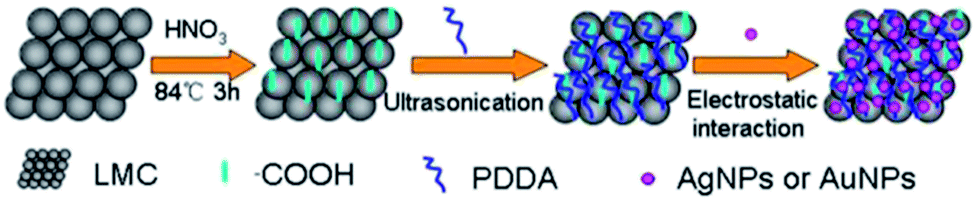 | ||
| Scheme 1 Procedure for the self-assembly of nanoparticles with PDDA-functionalized LMC. | ||
The electrostatic interactions between the PDDA–LMC and citrate-capped AgNPs and AuNPs were also investigated by zeta potential measurements and the results were summarized in Table 1. The mean zeta-potential of the PDDA–LMC was positive (+28.8 mV) because of the modification of cationic PDDA. When the negatively charged citrate-capped AgNPs (−26.4 mV) or AuNPs (−19.8 mV) were adsorbed on the PDDA–LMC, the zeta-potential of the PDDA–LMC was significantly altered. The zeta-potential of as-prepared AgNPs/PDDA–LMC and AuNPs/PDDA–LMC was +2.65 and +8.7 mV, respectively.
| Materials | PDDA–LMC | AgNPs | AuNPs | AgNPs/PDDA–LMC | AuNPs/PDDA–LMC |
|---|---|---|---|---|---|
| Zeta potential (mV) | +28.8 | −26.4 | −19.8 | +2.65 | +8.7 |
Fig. 1A shows the typical TEM images of the as-prepared LMC. It is obviously seen that a large amount of mesoporous are very uniform with alveolate surface. Fig. 1B and C show the typical images of the formation of AgNPs/PDDA–LMC and AuNPs/PDDA–LMC. It is found that all of the LMCs have been modified with high-density AgNPs and AuNPs. The corresponding histograms of particle size distribution were shown in Fig. S1A and B† with the mean particle diameter 20 ± 2.3 nm and 16 ± 2.2 nm for AgNPs and AuNPs. The self-assembly AgNPs and AuNPs were relatively homogenous except a few clusters consisting of several nanoparticles. These provided clear evidence to support the formation of AgNPs/PDDA–LMC or AuNPs/PDDA–LMC. Fig. 1D displays the wide-angle XRD pattern of PDDA–LMC (a), AgNPs/PDDA–LMC (b) and AuNPs/PDDA–LMC (c), respectively. The broad diffraction peaks at 2θ of 23° were observed for the three catalysts, corresponding to the (002) diffraction of the graphite. Several peaks of the AgNPs/PDDA–LMC (b) are characters of the Ag face-centered cubic (fcc) phase, namely the planes (111), (200), (220) and (311). The XRD peak positions of AuNPs/PDDA–LMC (c) are almost the same as those of the AgNPs/PDDA–LMC, indicating that the Ag and Au have almost the same crystal structures.
 | ||
| Fig. 1 Typical TEM images of (A) LMC, (B) AgNPs/PDDA–LMC, and (C) AuNPs/PDDA–LMC. (D) XRD patterns of (a) PDDA–LMC, (b) AgNPs/PDDA–LMC, and (c) AuNPs/PDDA–LMC. | ||
The successful synthesis of AgNPs/PDDA–LMC and AuNPs/PDDA–LMC was further confirmed by UV-vis absorption spectroscopy (Fig. S2†). The UV-vis spectra of AgNPs and AuNPs aqueous solution (curve a in Fig. S2A and B†) show a plasmonic band at 425 nm and 528 nm, respectively. After attachment onto the surface of the LMC (curve b in Fig. S2A and B†), both bands were shifted to 503 and 563 nm as a much wider peak. The red-shift of the surface-plasmonic band can be attributed to the interparticle interactions adsorbed on the coaxial nanocables as demonstrated by Giersig et al.36 No absorption peak can be observed in the LMC as shown in curve c of Fig. S2A and B.† All these results indicate that AgNPs and AuNPs have been decorated onto the LMCs.
Construction of AgNPs/PDDA–LMC-based H2O2 sensor
To testify the sensing application of the AgNPs/PDDA–LMC, we constructed an enzymeless H2O2 sensor by deposition of the aqueous dispersion of AgNPs/PDDA–LMC on a GCE surface. The influence of the different pH values of the buffer solution on the sensor was examined (Fig. S3†) and pH 6.5 was chosen for all experiments. Fig. 2 shows the cyclic voltammograms (CVs) of the bare GCE (dashed line), PDDA–LMC/GCE (dotted line), AgNPs/GCE (dash dotted line), and AgNPs/PDDA–LMC/GCE (solid line) in N2-saturated 0.2 M PBS at pH 6.5 in the presence of 1 mM H2O2 at a scan rate of 50 mV s−1. In the absence of H2O2, there was no reduction peak at AgNPs/PDDA–LMC/GCE (dash dotted dotted line). After addition of 1 mM H2O2, the bare GCE and PDDA–LMC/GCE electrodes only led a weak electrocatalytic reduction current, while AgNPs/GCE led an obvious reduction peak at −0.55 V. Compared with AgNPs/GCE, the AgNPs/PDDA–LMC/GCE has a similar peak current for the reduction of H2O2, but peak potential at AgNPs/PDDA–LMC/GCE (−0.45 V) is more positive than that at AgNPs/GCE. The more positive potential for H2O2 reduction indicates the significant electrocatalytic activity of AgNPs/PDDA–LMC. The enhanced electrochemical activity should be ascribed to integrated merit of LMC and high-loading and well-dispersed Ag nanoparticles. Well-dispersed Ag nanoparticles supplies more surface-active sites for the adsorption of reactants. | ||
| Fig. 2 CVs of the bare GCE, PDDA–LMC/GCE, AgNPs/GCE and AgNPs/PDDA–LMC/GCE in 0.2 M PBS at pH 6.5 in the presence of 1 mM H2O2 (scan rate: 50 mV s−1). | ||
Fig. 3 shows the typical amperometric current–time curve of AgNPs/PDDA–LMC/GCE with successive addition of H2O2 at the applied potential of −0.4 V. It ensures sufficient current response with lower background or less interference of other electroactive species in the solution.37 The response current dramatically increased after each addition of H2O2 and could attain to steady-state current in less than 2 s (inset a of Fig. 3). The inset b of Fig. 3 is the corresponding calibration plot between catalytic response and H2O2 concentration, which showed linear range of 0.02 mM to 9.62 mM (R = 0.9966) with an estimated detection limit of 6.5 μM based on signal to noise ratio of three. The sensitivity was calculated as 5.23 × 10−3 A mol−1 L−1. The detection limit was notably lower than those at the Ag nanoparticles-graphene-modified electrode (7 μM),38 the Hb/SAa-MWCNTsb-modified electrode (16.41 μM),39 LMC-modified electrode (44 μM)17 and the PEDOT/AgNPs-modified electrode (7 μM).40 The detailed comparison of this work with reported work regarding of the performance of the H2O2 sensor were summarized in Table S1.†
 | ||
| Fig. 3 Amperometric current–time curve of the AgNPs/PDDA–LMC/GCE with successive addition of H2O2 at the potential of −0.4 V. Inset shows the response time of the AgNPs/PDDA–LMC/GCE toward the addition of H2O2 (a) and the linear dependence of the current response with the H2O2 (b). | ||
Selectivity, reproducibility and stability of H2O2 sensor
The selectivity of the AgNPs/PDDA–LMC/GCE for the determination of H2O2 was also studied. Fig. 4A shows the current responses of the AgNPs/PDDA–LMC/GCE to several interfering substances, such as AA, UA, DA, AP, citric acid and glucose, which normally coexist with H2O2 in human blood. We observed an obvious current response with the addition of 0.1 mM H2O2. But no obvious current signal was observed with the addition of two times of AA, AP, DA, UA, citric and glucose, suggesting the good anti-interference ability of the sensor to the detection of H2O2 in real blood sample. | ||
| Fig. 4 (A) The amperometric response of the AgNPs/PDDA–LMC/GCE toward the addition of 0.1 mM H2O2, 0.2 mM UA, 0.2 mM AP, 0.2 mM DA, 0.2 mM AA, 0.2 mM glucose and 0.2 mM citric acid in 0.2 M PBS (pH 6.5). (B) The variation in the response current of H2O2 (1 mM) in 0.2 M PBS solution (pH 6.5) at the AgNPs/PDDA–LMC/GCE for 5 days. Scan rate: 50 mV s−1. | ||
Furthermore, the reproducibility of the sensor was also evaluated by testing the response of the sensor toward the addition of 1 mM H2O2 at a scan rate of 50 mV s−1. Based on the response current of one sensor to 1 mM H2O2, a relative standard deviation (RSD) of 4.2% was obtained for 5 successive measurements. Alternatively, the relative standard deviation (RSD) of 5.2% was obtained based on the responses of five sensors prepared independently toward 1 mM H2O2, These indicate the good reproducibility of the constructed sensor.
The long-term stability of the sensor was tested by periodical measurement of the response of the sensor to 1 mM H2O2 in PBS solution (pH 6.5) at a scan rate of 50 mV s−1. For the long-term stability test, we introduced Nafion (0.5 wt%) film as the protective layer of sensor and prepared Nafion/AgNPs/PDDA–LMC modified electrode. The sensor was stored at 4 °C when it was not used. As shown in Fig. 4B, one week later, the response current still remained 94.2%, suggesting the long-term stability of the sensor. Also, the operational stability was also examined. The Nafion/AgNPs/PDDA–LMC/GC electrode was used to determine 0.1 mM H2O2 for 60 min continuously (as Fig. S4†), and only 9% current diminutions at 60 min, indicating that the sensor is highly stable and can be successfully used for a long time.
Real sample analysis of H2O2 sensor
Based on the good selectivity, reproducibility and stability of the sensor, a practical applicability of the AgNPs/PDDA–LMC/GCE was tested for detection of H2O2 in human blood (from local hospital) by using standard addition method. A 50 μL (sample 1) or 100 μL (sample 2) aliquot of serum was added to 5 mL of 0.2 M PBS (pH = 6.5). Based on the current response, the concentration of H2O2 was determined utilizing the regression equation. In comparison with theoretical addition and determined concentration of H2O2, the recovery of H2O2 can be obtained. The results were listed in Table 2. These results confirmed the usefulness of the sensor in serum samples with practically application.| Sample | Original (μM) | Added (μM) | Found (μM) | Recover (%) |
|---|---|---|---|---|
| 1 | 0 | — | 23.95 | — |
| 23.95 | 50 | 72.38 | 97.9 | |
| 73.95 | 50 | 124.31 | 100.3 | |
| 2 | 0 | — | 23.88 | — |
| 23.88 | 100 | 124.1 | 100.2 | |
| 123.88 | 100 | 223.4 | 99.8 |
Direct electrocatalysis and amperometric detection of NaNO2 based on the AuNPs/PDDA–LMCs/GCE
To investigate the sensing application of the AuNPs/PDDA–LMC, a sensor for detecting nitrite oxidation was constructed by deposition of the aqueous dispersion of the AuNPs/PDDA–LMC on a GCE surface. By testing the amperometric response of the sensor toward the addition of 10 mM NaNO2 into 0.2 M PBS with different pH values, we found that the response current was the largest at pH 6.5 (as shown in Fig. S5†). Therefore, all experiments were performed in PBS at pH 6.5. Fig. 5 shows the CVs of the bare GCE (dashed line), PDDA–LMC/GCE (dotted line), AuNPs/GCE (dash dotted line) and AuNPs/PDDA–LMC/GCE (solid line) in 0.2 M PBS at pH 6.5 in the presence of 10 mM NaNO2. Although a well-defined oxidation peak of nitrite could be observed at all electrodes, the catalytic current was dramatically larger at the AuNPs/PDDA–LMC/GCE than that of at other electrodes. Moreover, the current peak showed a 0.16 V negative shift at the AuNPs/PDDA–LMC/GCE (solid line). These indicate a better electrocatalytic activity of AuNPs/PDDA–LMC/GCE (solid line). | ||
| Fig. 5 CVs of the bare GCE, PDDA–LMC/GCE, AuNPs/GCE and AuNPs/PDDA–LMC/GCE in 0.2 M PBS at pH 6.5 in the presence of 10 mM NaNO2 (scan rate: 50 mV s−1). | ||
Fig. 6 shows the amperometric i–t curves at the AuNPs/PDDA–LMC/GCE with successive additions of nitrite at +0.8 V. The response current gradually increased after each addition of nitrite into the stirring PBS, and reached the maximum steady-state current within 2 s. Inset b of Fig. 6 shows the corresponding calibration plot with the linear detection range from 5 μM to 7.24 mM (R = 0.998) and the estimated detection limit of 0.42 μM could be obtained at a signal-to-noise ratio of 3. The detection limit was even lower than that of at the noble metal nanoparticles-modified electrode, such as at the Pt/Cha/GCE (0.47 μM),40 the Hb/SAa-MWCNTsb modified electrode (16.41 μM),41 and the nano-Ptf/P3MTg/GCE (1.5 μM),42 indicating the enhanced sensitivity of the sensor to the detection of NaNO2. The performance of the AuNPs/PDDA–LMC/GCE for the detection of nitrate is even better than those of carbon nanotube,43–45 graphene oxide,46 and carbon nanofibre/hemin-based47 nanomaterials. The detailed comparisons of the performance of the sensor with others in the detection of NaNO2 were summarized in the Table S2.†
 | ||
| Fig. 6 The amperometric response of the AuNPs/PDDA–LMC/GCE with successive additions of NaNO2 at the potential of +0.8 V. Inset (a) shows the linear dependence of the current response with the NaNO2 and (b) shows the amplified response curve at low concentration of NaNO2. | ||
Selectivity, reproducibility and stability of NaNO2 sensor
The interference of some common chemical species on the response current of the constructed sensor toward the addition of 0.2 mM NaNO2 was also studied. It can be seen that the sensor shows no detected signal upon the addition of Na2SO4, KCl, NH4NO3, Na2CO3, glucose, and C2H5OH and the presence of these chemicals also has no obvious influence on the detection of NaNO2 at the AuNPs/PDDA–LMC/GCE (Fig. 7A), suggesting the good selectivity of the constructed sensor toward the detection of NaNO2. The reproducibility of the sensor was also investigated by current–time method for seven repetitive measurements with addition of 10 mM NaNO2. The relative standard deviation (RSD) of the sensitivity was less than 4.1%.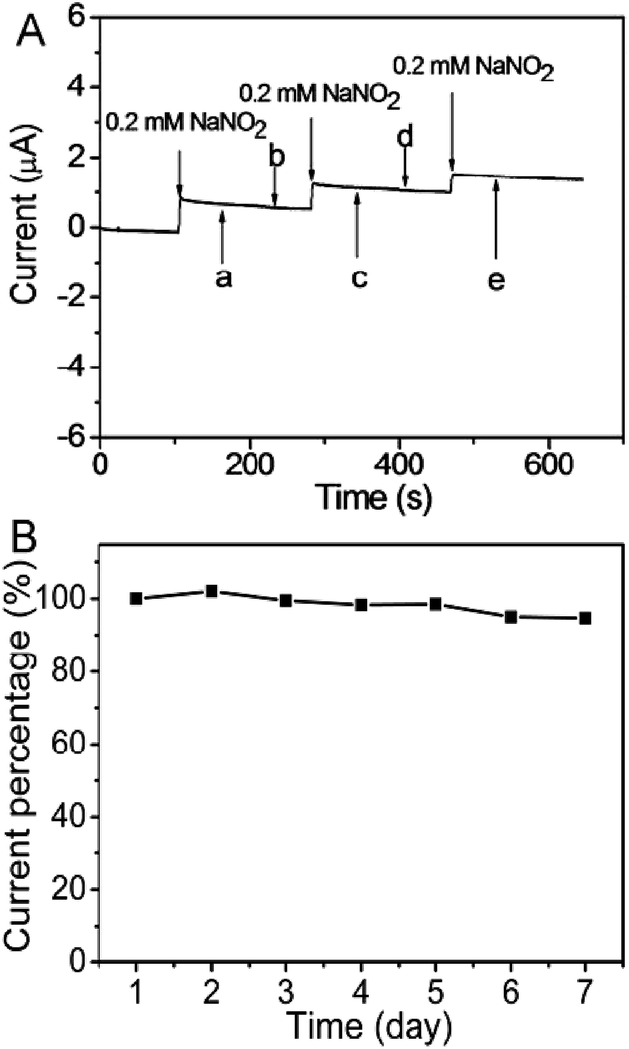 | ||
| Fig. 7 (A) The amperometric response of 0.2 mM NaNO2 at the AuNPs/PDDA–LMC/GCE in the presence of various interfering chemicals such as 2 mM (a) MgSO4, (b) NH4Cl, (c) Na2CO3, (d) KCl, (e) glucose and (f) C2H5OH in 0.2 M PBS (pH 6.5). (B) The variation in the response current of (10 mM) NaNO2 in 0.2 M PBS solution (pH 6.5) at the AuNPs/PDDA–LMC/GCE for 5 days. Scan rate: 50 mV s−1. | ||
The stability of the sensor was periodically investigated by amperometric measurements in the presence of 10 mM NaNO2. When it was not in use, the Nafion/AuNPs/PDDA–LMC/GCE was stored under dry conditions at 4 °C for one week. As shown in Fig. 7B, the deviation after one week storage was 5.3% in comparison with its original value. Also, only 12% current diminutions at 60 min was observed in determination of 1.0 mM NaNO2 (as Fig. S6†). The results indicated the good long-term stability of the sensor.
Conclusions
In this work, we presented a facile and simple self-assembly process to prepare AgNPs/PDDA–LMC and AuNPs/PDDA–LMC nanocomposites. The cationic PDDA was used as a building bulk, which reacted with the citrate-capped AgNPs or AuNPs by electrostatic interaction. The nanocomposites integrated the high-efficient catalysis of AgNPs or AuNPs with the beneficial structure of LMC. Therefore, the AgNPs/PDDA–LMC/GCE showed good eletrocatalysts activity for the detection of hydrogen peroxide and the AuNPs/PDDA–LMC/GCE showed higher eletrocatalysts activity for the detection of sodium nitrite with lower detection limit and good selectivity, reproducibility and stability. Such nanocomposites provide a promising platform for the preparation of other sensors.Acknowledgements
This work was financially supported by the President Funds of the Chinese Academy of Sciences and the Natural Science Foundation of Jilin Province (201215092).Notes and references
- Y. Wan, Y. Shi and D. Zhao, Chem. Mater., 2007, 20, 932 CrossRef.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743 CrossRef CAS.
- M. Zhou, J. Guo, L. Guo and J. Bai, Anal. Chem., 2008, 80, 4642 CrossRef CAS PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712 CrossRef CAS.
- A. Vinu, M. Miyahara and K. Ariga, J. Phys. Chem. B, 2005, 109, 6436 CrossRef CAS PubMed.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316 CrossRef CAS PubMed.
- K. Li, Y. Luo, Z. Yu, M. Deng, D. Li and Q. Meng, Electrochem. Commun., 2009, 11, 1346 CrossRef CAS PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937 CrossRef CAS.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. Bum Kim, Chem. Commun., 1999, 2177 RSC.
- M. Zhou, L. Shang, B. Li, L. Huang and S. Dong, Biosens. Bioelectron., 2008, 24, 442 CrossRef CAS PubMed.
- Z. Tan, H. Xiao, R. Zhang, Z. Zhang and S. Kaliaguine, New Carbon Mater., 2009, 24, 333 CrossRef CAS.
- L. Sterk, J. Górka, A. Vinu and M. Jaroniec, Microporous Mesoporous Mater., 2012, 156, 121 CrossRef CAS PubMed.
- Y. Zhang, X. Bo, C. Luhana and L. Guo, Electrochim. Acta, 2011, 56, 5849 CrossRef CAS PubMed.
- X. Bo, J. Bai, J. Ju and L. Guo, Anal. Chim. Acta, 2010, 675, 29 CrossRef CAS PubMed.
- L. Wang, J. Bai, X. Bo, X. Zhang and L. Guo, Talanta, 2011, 83, 1386 CrossRef CAS PubMed.
- X. Bo, J. Bai, B. Qi and L. Guo, Biosens. Bioelectron., 2011, 28, 77 CrossRef CAS PubMed.
- J. Bai, B. Lu, X. Bo and L. Guo, Electrochem. Commun., 2010, 12, 1563 CrossRef CAS PubMed.
- H. Wang, X. Bo, J. Bai, L. Wang and L. Guo, J. Electroanal. Chem., 2011, 662, 281 CrossRef CAS PubMed.
- X. Yan, D. Pan, H. Wang, X. Bo and L. Guo, J. Electroanal. Chem., 2011, 663, 36 CrossRef CAS PubMed.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836 CrossRef.
- A. J. Wain, Electrochim. Acta, 2013, 92, 383 CrossRef CAS PubMed.
- Y. Luo, W. Lu, G. Chang, F. Liao and X. Sun, Electrochim. Acta, 2011, 56, 8371 CrossRef CAS PubMed.
- M. Yang, Y. Yang, Y. Liu, G. Shen and R. Yu, Biosens. Bioelectron., 2006, 21, 1125 CrossRef CAS PubMed.
- J. Yin, X. Qi, L. Yang, G. Hao, J. Li and J. Zhong, Electrochim. Acta, 2011, 56, 3884 CrossRef CAS PubMed.
- Y. Fang, S. Guo, C. Zhu, Y. Zhai and E. Wang, Langmuir, 2010, 26, 11277 CrossRef CAS PubMed.
- A. Schwake, B. Ross and K. Cammann, Sens. Actuators, B, 1998, 46, 242 CrossRef CAS.
- Y. Wang, J. Huang, C. Zhang, J. Wei and X. Zhou, Electroanalysis, 1998, 10, 776 CrossRef CAS.
- J. Lee and J. D. Helmann, Nature, 2006, 440, 363 CrossRef CAS PubMed.
- M. A. Yorek, Free Radical Res., 2003, 37, 471 CrossRef CAS.
- S. Y. Ha and S. Kim, J. Electroanal. Chem., 1999, 468, 131 CrossRef CAS.
- A. L. Sousa, W. J. R. Santos, R. C. S. Luz, F. S. Damos, L. T. Kubota, A. A. Tanaka and S. M. C. N. Tanaka, Talanta, 2008, 75, 333 CrossRef CAS PubMed.
- B. Xu, L. Peng, G. Wang, G. Cao and F. Wu, Carbon, 2010, 48, 2377 CrossRef CAS PubMed.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391 CrossRef CAS.
- G. Zhou and W. Wang, Orient. J. Chem., 2012, 28, 651 CrossRef CAS.
- Y. Wang, H. Wei, B. Li, W. Ren, S. Guo, S. Dong and E. Wang, Chem. Commun., 2007, 5220 RSC.
- M. A. Correa-Duarte, N. Sobal, L. M. Liz-Marzán and M. Giersig, Adv. Mater., 2004, 16, 2179 CrossRef CAS.
- B. Wolfrum, M. Zevenbergen and S. Lemay, Anal. Chem., 2008, 80, 972 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, L. Wang, X. Qin, J. Tian, W. Lu, G. Chang and X. Sun, RSC Adv., 2012, 2, 538 RSC.
- H. Y. Zhao, W. Zheng, Z. X. Meng, H. M. Zhou, X. X. Xu, Z. Li and Y. F. Zheng, Biosens. Bioelectron., 2009, 24, 2352 CrossRef CAS PubMed.
- S. Wang, Y. Yin and X. Lin, Electrochem. Commun., 2004, 6, 259 CrossRef CAS PubMed.
- N. Zhu, Q. Xu, S. Li and H. Gao, Electrochem. Commun., 2009, 11, 2308 CrossRef CAS PubMed.
- Y. Zhou, H. Xian, F. Li, S. Wu, Q. Lu, Y. Li and L. Wang, Electrochim. Acta, 2010, 55, 5905 CrossRef CAS PubMed.
- Y. Zhang, R. Yuan, Y. Q. Chai, W. J. Li, X. Zhong and H. A. Zhong, Biosens. Bioelectron., 2011, 26, 3977 CrossRef CAS PubMed.
- A. Afkhami, T. Madrakian, H. Ghaedi and H. Khanmohammadi, Electrochim. Acta, 2012, 66, 255 CrossRef CAS PubMed.
- D. Gligor and A. Walcarius, J. Solid State Electrochem., 2014, 18, 1519 CrossRef CAS.
- V. Mani, A. P. Periasamy and S.-M. Chen, Electrochem. Commun., 2012, 17, 75 CrossRef CAS PubMed.
- F. Valentini, L. Cristofanelli, M. Carbone and G. Palleschi, Electrochim. Acta, 2012, 63, 37 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The characterization of AgNPs/PDDA–LMC and AuNPs/PDDA–LMC, additional electrochemical investigations. See DOI: 10.1039/c4ra14374d |
| This journal is © The Royal Society of Chemistry 2015 |