
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unexpected formation of [Ru(η5-C5H5)(PH{CH2N(CH2CH2)2O}2)(PPh3)2]BF4 – the first “piano-stool” ruthenium complex bearing a secondary aminomethylphosphane ligand†
Michał
Płotek
ab,
Radosław
Starosta
*c,
Urszula K.
Komarnicka
c,
Agnieszka
Skórska-Stania
a,
Grażyna
Stochel
a,
Agnieszka
Kyzioł
*a and
Małgorzata
Jeżowska-Bojczuk
c
aFaculty of Chemistry, Jagiellonian University in Krakow, Ingardena 3, 30-060 Krakow, Poland. E-mail: kyziol@chemia.uj.edu.pl
bFaculty of Conservation and Restoration of Works of Art, Jan Matejko Academy of Fine Arts in Krakow, Lea 27-29, 30-052 Krakow, Poland
cFaculty of Chemistry, University of Wroclaw, Joliot-Curie 14, 50-383 Wroclaw, Poland. E-mail: radoslaw.starosta@chem.uni.wroc.pl
First published on 2nd December 2014
Abstract
In this paper we report the reaction of [Ru(η5-C5H5)Cl(PPh3)2] with P{CH2N(CH2CH2)2O}3 in the presence of NaBF4, in which, apart from the Cl− substitution, an unexpected P–C bond cleavage in the tertiary phosphane is observed. It results in the formation of [Ru(η5-C5H5)(PH{CH2N(CH2CH2)2O}2)(PPh3)2]BF4 (1) – the first “piano-stool” ruthenium complex with a secondary aminomethylphosphane ligand.
Metal complexes have been investigated as potential chemotherapeutic agents during the past few decades.1 The discovery of therapeutic activity of (ImH)[trans-RuCl4(DMSO)Im] (NAMI-A) and (IndH)[trans-RuCl4(Ind)2] (KP1019) allowed the ruthenium complexes to achieve status of promising candidates for novel cancer therapy.2 The ruthenium ion in both complexes has an oxidation state of +3. Nevertheless, it is suggested that reduction of Ru(III) to Ru(II) takes place inside the cell prior to DNA binding, which is known as the activation by reduction mechanism.3 The hypothesis that ruthenium(II) agents are much more active factors in biological system than their ruthenium(III) analogues caused that more attention has been paid in the investigations of Ru(II) coordination compounds. Nowadays, ruthenium(II) compounds, especially “piano-stool” [Ru(arene) (L1)(L2)(L3)] complexes, are one of the most explored groups of potential drugs.4 Moreover, the easy introduction of different L ligands (e.g. halides, phosphanes or amines) enables significant modifications of not only stability and physicochemical properties but also biological activity of these compounds.
In recent years, we have been interested in photochemical and biological properties of ruthenium compounds.5 Currently, we are focused on the synthesis of novel “piano-stool” ruthenium complexes. Our preliminary goal was to substitute of triphenylphosphane molecules or chloride ion in [Ru(η5-C5H5)Cl(PPh3)2] complex, as it was shown by Ashby et al.6 For the Cl− replacement we chose a method presented by Romerosa et al.7 involving the presence of sodium tetrafluoroborate. As the replacing ligands we decided to use tris(aminomethyl)phosphanes. These ligands are the large and interesting class of phosphanes for several reasons. Firstly, they are stable in water solution and, to some extent, in the presence of oxygen.8 Secondly, these ligands can be easily functionalised. For example, aminomethylphosphanes derived from amino acids9 or prepared from the highly water-soluble aliphatic secondary amines,10 seem to be interesting in terms of the formation of potential conjugates with a wide range of biomolecules. Moreover, in our previous papers we presented a group of copper(I) complexes with tris(aminomethyl)phosphanes derived from morpholine (P{CH2N(CH2CH2)2O}3) or piperazine (P{CH2–N(CH2CH2)2N–R}3).8 Promising in vitro cytotoxicity of these complexes encouraged us to extend our studies to ruthenium compounds.
Herein, we report the reaction of [Ru(η5-C5H5)Cl(PPh3)2] with P{CH2N(CH2CH2)2O}3 (P(CH2-morph)3) in the presence of NaBF4, which did not lead to a straightforward substitution of the Cl− ion, but involved a simultaneous P–C bond cleavage in P(CH2-morph)3, resulting in formation of the secondary phosphane – PH(CH2-morph)2, which gave the [Ru(η5-C5H5){PH(CH2-morph)2}(PPh3)2]BF4 complex (1) (Fig. 1).
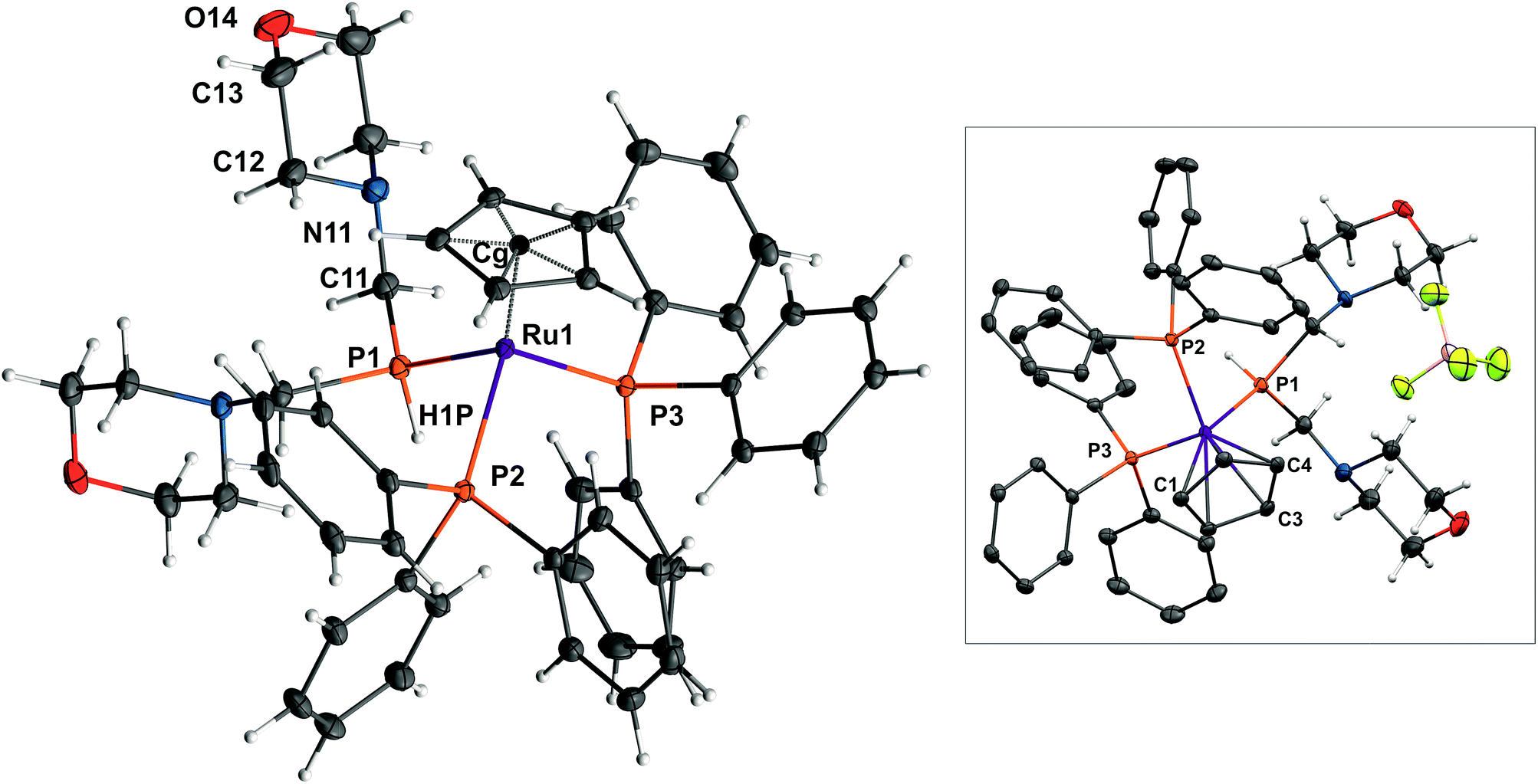 | ||
| Fig. 1 Two views of the crystal structure of complex 1 (30% probability ellipsoids). Selected bond distances (Å) and angles (°): Ru(1)–C(1) = 2.250(2), Ru(2)–C(2) = 2.238(2), Ru(1)–C(3) = 2.244(2), Ru(1)–C(4) = 2.255(2), Ru(1)–C(5) = 2.253(2), Ru(1)–CgCp (cyclopentadienyl centre of gravity) = 1.894(2), Ru(1)–P(1) = 2.294(1), Ru(1)–P(2) = 2.366(1), Ru(1)–P(3) = 2.335(1), P(1)–Ru(1)–P(2) = 89.71(2), P(1)–Ru(1)–P(3) = 92.94(2), P(2)–Ru(1)–P(3) = 102.08(2), CgCp–Ru(1)–P(1) = 126.54(3), CgCp–Ru(1)–P(2) = 121.71(3), CgCp–Ru(1)–P(3) = 117.14(3). | ||
Addition of P(CH2-morph)3 to the orange suspension of [Ru(η5-C5H5)Cl(PPh3)2] and NaBF4 led to the formation of yellow solution after 3 hours of reflux.‡31P{1H} NMR spectrum of this solution (see ESI Fig. S3†) showed that the main product possess three phosphorus atoms – two equivalent atoms observed as a doublet and the other one, which is observed as a triplet. Such a spectrum could suggest a simple replacement of the chloride ion by the tris(aminomethyl)phosphane. However, the analysis of the crystal structure of this complex revealed that, in time of synthesis, tertiary phosphine P(CH2-morph)3 was rearranged to secondary PH(CH2-morph)2. 1 is therefore a “piano-stool” complex, in which chloride was substituted by the secondary aminomethylphosphane.
A large number of ruthenium complexes with secondary and/or primary phosphanes were synthesized until now.11 However, the cleavage of the phosphane P–C bond during complex formation is unprecedented not only for the aminomethylphosphanes. A similar reaction was observed by Higham et al.12 only for two hydroxymethylphosphanes. They reported analogous process when fourfold excess of P(CH2OH)3 or P(CH2OH)2Ph was mixed with RuCl3·H2O. As a result, they obtained the coordination compounds possessing two molecules of the tertiary phosphane and two molecules of the secondary one: [Ru{PPh(CH2OH)2}2{PPh(CH2OH)H}2Cl2] and [Ru{P(CH2OH)3}2{P(CH2OH)2H}2Cl2].
The complex 1 crystallizes in tetragonal crystal system; space group I41/a. The asymmetric unit contains sixteen equivalent [Ru(η5-C5H5){PH(CH2-morph)2}(PPh3)2]+ cations and tetrafluoroborate anions. Replacement of chloride with the PH(CH2-morph)2 leads to a slight elongation of one of the Ru-PPh3 bonds compared to the parent complex.13 The bond lengths are equal: Ru(1)–P(3) = 2.335(1) Å, Ru(1)–P(2) = 2.366(1) Å for 1 and Ru–P(1) = 2.337(1), Ru–P(2) = 2.335(1) Å for [Ru(η5-C5H5)Cl(PPh3)2]. Moreover, the average distance from the carbon atoms of C5H5− ligand to Ru(1) central atom, equal to 2.248(2) Å, is also longer than for [Ru(η5-C5H5)Cl(PPh3)2] (2.207(3) Å). The angle between Ru–PPh3 bonds found for 1 (P(2)–Ru(1)–P(3) = 102.08(2)°) is smaller than for [Ru(η5-C5H5)Cl(PPh3)2] (P(1)–Ru–P(2) = 103.99(4)°). Above data prove steric hindrances in 1 caused by the presence of PH(CH2-morph)2 ligand and may be related to a facilitation of the –CH2-morph group release.
Measurement of 31P NMR spectrum confirmed the presence of the secondary phosphane. The upfield signal of PH(CH2-morph)2, which is observed as a triplet at −12.96 ppm (2J(P1–P2,3) = 41.2 Hz) in the proton-decoupled 31P{H} NMR spectrum, in 31P NMR spectrum appears as a doublet of triplets (Fig. 2). Appearance of a new, large coupling (338.6 Hz) proves that phosphorous atom is directly bound with hydrogen atom. Integration of the signals in the proton spectrum also confirms that aminomethylphosphane loses one –CH2-morph moiety during of complex preparation. The PPh3 part of the 13C{H} spectrum of 1, similarly to the spectrum of [Ru(η5-C5H5)Cl(PPh3)2], consists of four signals: the multiplet of the Cipso atoms, the singlet of Cpara atoms and two pseudotriplets of the Corto and Cmeta atoms (see ESI Fig. S6 and S8†). The latter signals arise from a second-order effect, referred to as “virtual coupling”,14 observed usually for the phosphanes in trans positions to one another. The PH(CH2-morph)2 part consists of a doublet of C11 carbon atoms with the 1J(C–P) = 36.1 Hz – much larger than observed for the free P(CH2-morph)3 ligand, a doublet of C12 and a singlet of C13 atoms.
 | ||
| Fig. 2 Comparison of the 31P and 31P{1H} NMR spectra (CDCl3, 300 K) of complex 1. | ||
The other data are fully consistent with the crystal structure of 1.‡ In the infrared spectrum the band assigned to P–H stretching is observed at 2346 cm−1. The location of the band at lower wavenumber in comparison with that of [Ru{P(CH2OH)3}2{PH(CH2OH)2}2Cl2] (2374 cm−1)12 suggests that P–H bond in 1 is noticeably weaker. It is worth to mention that 1 is stable under normal conditions (the air and the presence of moisture). Fragmentation of the [Ru(η5-C5H5){PH(CH2-morph)}(PPh3)2]+ ion in ESI-MS takes place only in a small degree, even under acidic conditions. Among products of fragmentation it is possible to identify [Ru(η5-C5H5){PH2(CH2-morph)}(PPh3)2]+ and [Ru(η5-C5H5)(PPh3)2]+ ions. It suggests that –CH2-morph moiety as well as secondary phosphane PH(CH2-morph)2 are more susceptible to elimination than triphenylphosphane parts of complex 1.
Conclusions
The synthesized complex, to the best of our knowledge, is a first metal complex bearing a secondary bis(aminomethyl)phosphane ligand. The presented formation of ruthenium(II) complex containing a reactive P–H bond, offers a great scope for further ligand functionalization. Moreover, our recent studies proved that discussed process is not an individual case. The preliminary experiments showed that similar reaction takes place in the case of other tris(aminomethyl)phosphanes as well as indenyl or pentamethylcyclopentadienyl “piano-stool” starting complexes. Now we are focused on elucidation of the mechanism of the phosphane P–C bond cleavage as well as the biological properties of the resulting complex.Notes and references
- Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine, ed. M. Gielen and E. R. Tiekink, Wiley, 2005 CrossRef CAS; M. A. Hannon, Pure Appl. Chem., 2007, 79, 2243 CrossRef CAS.
- G. Suss-Fink, Dalton Trans., 2010, 39, 1673 RSC.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198 CrossRef CAS PubMed.
- L. K. Filak, D. S. Kalinowski, T. J. Bauer, D. R. Richardson and V. B. Arion, Inorg. Chem., 2014, 53, 6934 CrossRef CAS PubMed; R. Pettinari, C. Pettinari, F. Marchetti, B. W. Skelton, A. H. White, L. Bonfili, M. Cuccioloni, M. Mozzicafreddo, V. Cecarini, M. Angeletti, M. Nabissi and A. M. Eleuteri, J. Med. Chem., 2014, 57, 4532 CrossRef PubMed; C. M. Clavel, E. Paunescu, P. Nowak-Sliwinska, A. W. Griffioen, R. Scopelliti and P. J. Dyson, J. Med. Chem., 2014, 57, 3546 CrossRef PubMed; J. Kljun, A. J. Scott, T. Lanisnik Rizner, J. Keiser and I. Turel, Organometallics, 2014, 33, 1594 CrossRef; K. Dralle Mjos and Ch. Orvig, Chem. Rev., 2014, 114, 4540 CrossRef PubMed; A. Grau-Campistany, A. Massaguer, D. Carrion-Salip, F. Barragan, G. Artigas, P. Lopez-Senin, V. Moreno and V. Marchan, Mol. Pharm., 2013, 10, 1964 CrossRef PubMed; C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677 CrossRef; T. Kuster, N. Lense, F. Barna, A. Hemphill, M. K. Kindermann, J. W. Heinicke and C. A. Vock, J. Med. Chem., 2012, 55, 4178 CrossRef PubMed.
- M. Brindell, E. Kuliś, S. K. C. Elmoroth, K. Urbańska and G. Stochel, J. Med. Chem., 2005, 48, 7298 CrossRef CAS PubMed; M. Brindell, D. Piotrowska, A. A. Shoukry, G. Stochel and R. Van Eldik, JBIC, J. Biol. Inorg. Chem., 2007, 12, 809 CrossRef PubMed; M. Brindell, I. Stawoska, J. Supel, A. Skoczowska, G. Stochel and R. Van Eldik, JBIC, J. Biol. Inorg. Chem., 2008, 13, 909 CrossRef PubMed; O. Mazuryk, K. Kurpiewska, K. Lewiński, G. Stochel and M. Brindell, J. Inorg. Biochem., 2012, 116, 11 CrossRef PubMed; M. Oszajca, E. Kuliś, G. Stochel and M. Brindell, New J. Chem., 2014, 38, 3386 RSC; O. Mazuryk, M. Maciuszek, G. Stochel, F. Suzenet and M. Brindell, J. Inorg. Biochem., 2014, 134, 83 CrossRef PubMed.
- G. S. Ashby, M. I. Bruce, I. B. Tomkins and R. C. Wallis, Aust. J. Chem., 1979, 32, 1003 CrossRef CAS.
- A. Romerosa, M. Saoud, T. Campos-Malpartida, C. Lidrissi, M. Serrano-Ruiz, M. Peruzzini, J. A. Garrido and F. Garcia-Maroto, Eur. J. Inorg. Chem., 2007, 18, 2803 CrossRef.
- R. Starosta, M. Florek, J. Król, M. Puchalska and A. Kochel, New J. Chem., 2010, 34, 1441 RSC; R. Starosta, K. Stokowa, M. Florek, J. Król, A. Chwiłkowska, J. Kulbacka, J. Saczko, J. Skała and M. Jeżowska-Bojczuk, J. Inorg. Biochem., 2011, 105, 1102 CrossRef CAS PubMed; R. Starosta, M. Puchalska, J. Cybińska, M. Barys and A.-V. Mudring, Dalton Trans., 2011, 40, 2459 RSC; R. Starosta, A. Bykowska, A. Kyzioł, M. Płotek, M. Florek, J. Król and M. Jeżowska-Bojczuk, Chem. Biol. Drug Des., 2013, 82, 579 Search PubMed.
- C. Abu-Gnim and I. Amer, J. Organomet. Chem., 1996, 516, 235 CrossRef CAS; F. P. Pruchnik and P. Smolenski, Pol. J. Chem., 2007, 81, 1771 Search PubMed; K. Raghuraman, K. K. Katti, L. J. Barbour, N. Pillarsetty, C. L. Barnes and K. V. Katti, J. Am. Chem. Soc., 2003, 125, 6955 CrossRef PubMed; D. E. Berning, K. V. Katti, Ch. L. Barnes and W. A. Volkert, J. Am. Chem. Soc., 1999, 121, 1658 CrossRef.
- H. Coates and P. A. T. Hoye, Br. Pat., 842593, 1960; H. Coates and P. A. T. Hoye, US Pat., 3035053, 1962.
- S. I. M. Paris, F. R. Lemke, R. Sommer, P. Lonnecke and E. Hey-Hawkins, J. Organomet. Chem., 2005, 690, 1807 CrossRef CAS PubMed; S. I. M. Paris, J. F. Petersen, E. Hey-Hawkins and M. P. Jensen, Inorg. Chem., 2006, 45, 5561 CrossRef PubMed; A. J. Blake, N. R. Champness, R. J. Forder, Ch. S. Frampton, C. A. Frost, G. Reid and R. H. Simpson, J. Chem. Soc., Dalton Trans., 1994, 3377 RSC.
- L. Higham, A. K. Powell, M. K. Whittlesey, S. Wocadlo and P. T. Wood, Chem. Commun., 1998, 1107 RSC; L. J. Higham, M. K. Whittlesey and P. T. Wood, Dalton Trans., 2004, 4202 RSC.
- M. I. Bruce, F. S. Wong, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1981, 1398 RSC.
- D. W. Krassowski, J. H. Nelson, K. R. Brower, D. Hauenstein and R. A. Jacobson, Inorg. Chem., 1988, 27, 4294 CrossRef CAS.
- CrysAlisPRO, Oxford Diffraction Agilent Technologies UK Ltd, Yarnton, England Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic and crystal data. CCDC 1023104. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13037e |
| ‡ Preparation: [Ru(η5-C5H5)Cl(PPh3)2] were bought in Stream Chemicals, whereas other chemicals were from Sigma-Aldrich. P(CH2-morph)3 was synthesised according to literature procedure.7 Each synthetic step was performed in inert atmosphere: [Ru(η5-C5H5)Cl(PPh3)2] (0.116 g, 0.16 mmol) and sodium tetrafluoroborate (0.026 g, 0.24 mmol) were refluxed in methanol by 20 minutes. After this time the orange suspension was cooled to room temperature, solid aminomethylphosphane P(CH2-morph)3 (0.080 g, 0.24 mmol) was added and the mixture was refluxed one more time by 3 hours. Obtained yellow solution was evaporated to 1 ml and biphasic mixture of diethyl ether and water was added. Immediately yellow precipitate appeared at the interface. The precipitate was washed by water, methanol and diethyl ether and dried under vacuum. Yield: 65%. Anal. Found: C, 60.25; H, 5.55; N, 2.78%. Anal. Calc. for RuC51H56N2O2P3BF4: C, 60.66; H, 5.59; N, 2.77%. Spectroscopic data: 31P{1H} NMR (CDCl3) δ(ppm), (J [Hz]): −12.96 (t, 41.2), 42.03 (d, 41.2); 31P NMR (CDCl3) δ(ppm), (J [Hz]): −12.96 (d-t, 41.2 and 338.6), 42.03 (d, 41.2); 1H NMR (CDCl3) δ(ppm), (J [Hz]): 2.28 (m, 8 H), 2.94 (m, 4 H), 3.62 (m, 8 H), 4.95 (s, 5 H), 6.94 (bt, 8.6, 12 H), 7.33 (t, 7.2, 12 H), 7.45 (t, 7.2, 6 H); 13C NMR (CDCl3) δ(ppm), (J [Hz]): 54.31 (d, 5.5), 55.97 (d, 36.1), 66.95 (s), 85.42 (s), 128.52 (bt, 4.1), 130.42 (s), 133.56 (bt, 4.8), 136.41 (m); IR (ATR) (vmax [cm−1]): 3103w, 3054w, 2954w, 2841w, 2799w, 2346w, 1480w, 1451w, 1433m, 1304w, 1285m, 1256w, 1233w, 1205w, 1186w, 1159w, 1113m, 1085m, 1058s, 1018s, 1000m, 987m, 926w, 914w, 900m, 875m, 864s, 847m, 818m, 802w, 788w, 752s, 693s, 610w, 590w, 533m, 516s, 487s, 456m, 430m; +ESI-MS (MeOH) (m/z): 953.27 (2.34%), 923.26 (100%), 824.19 (1.99%), 426.13 (1.65%); +ESI-MS (MeOH/HCOOH) (m/z): 1011.27 (3.32%), 953.27 (1.08%), 923.26 (32.84%), 824.19 (1.43%), 691.12 (2.58%), 462.13 (100%), 263.10 (3.39%), 233.14 (6.38%); Yellow crystals suitable for the X-ray analysis were obtained by slow diffusion of diethyl ether to methanolic solution. Crystal data: [Ru(η5-C5H5)(PH{CH2N(CH2CH2)2O}2) (PPh3)2]BF4 (1) ≡ C51H56BF4N2O2P3Ru, M = 1009.77 g mol−1, crystal size: 0.28 × 0.25 × 0.15 mm, crystal system: tetragonal, space group: I41/a, a = 28.1859(1) Å, b = 28.1859(1) Å, c = 23.7177(2) Å, α = β = γ = 90°; V = 18 Instruments: NMR spectra were registered using Bruker Avance III 600 MHz spectrometer in CDCl3 with traces of CHCl3 as an internal reference for 1H and 13C and 85% H3PO4 in H2O as an external standard for 31P. The infrared spectrum was recorded using Bruker Alpha FT-IR spectrometer. All spectra were collected using the Platinum-ATR-sampling module equipped with diamond crystal. 64 interferograms were recorded with a resolution of 4 cm−1, in the range of 4000 cm−1 to 400 cm−1. A mass spectrometer with a time of flight mass analyzer (MicrOTOF-Q II, Bruker, Germany) was used. ESI was used as the ion source with conditions as follows: nebulizer pressure: 0.4 bar, dry gas: 4.0 l min−1 heated to 180 °C. Data were recorded in the positive ion mode and profile spectra were acquired in the mass range 50–3000 m/z. End plate offset was – 500 V and capillary voltage: 4500 V. Mass resolving power of the instrument was over 18 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 842.4(2) Å3, Dcalc(
842.4(2) Å3, Dcalc(| This journal is © The Royal Society of Chemistry 2015 |