DOI:
10.1039/C4RA14184A
(Paper)
RSC Adv., 2015,
5, 12346-12353
Nanosize α-Bi2O3 decorated Bi2MoO6 via an alkali etching process for enhanced photocatalytic performance†
Received
9th November 2014
, Accepted 13th January 2015
First published on 15th January 2015
Abstract
Bi2MoO6/Bi2O3 heterojunction photocatalysts were synthesized through a facile alkali etching method and their photocatalytic activity was evaluated by photodegradation of Rhodamine B (RhB) under visible-light irradiation. The photocatalysts were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM)/transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and UV-visible diffuse reflectance spectroscopy. XRD, TEM and XPS results confirm that the Bi2MoO6/Bi2O3 heterojunctions were formed after the alkali etching process. The photocatalytic activity can be easily controlled by adjusting the etching time and the highest visible-light-responsive photocatalytic performance was observed on Bi2MoO6/Bi2O3 composite that was etched in 0.1 M NaOH for 45 min. Moreover, the mechanistic study suggested that the photoinduced holes played a major role for the RhB degradation and the photocurrent tests confirmed that the effective electron–hole separation on the interface of the p–n junction promotes the photocatalytic process.
1. Introduction
Semiconductor photocatalysts have attracted increasing interest for their potential applications in global environmental pollutant control, hydrogen production and the degradation of organic/organoarsenic compounds.1–4 Up to now, a large number of semiconductor photocatalysts have been intensively investigated because of their physical and chemical properties, especially their ability to decompose organic dyes.5 However, most semiconductor photocatalysts (like TiO2, ZnO) can be only excited under UV irradiation which accounts for less than 5% of sunlight.6–8 In order to efficiently utilize the visible light emission that occupies 43% of the solar spectrum, the development of visible-light-responsive photocatalysts is urgently needed.
Recently, the bismuth-based photocatalytic materials, such as Bi2WO6, Bi2O3, Bi2O2CO3, BiOX (X = I, Cl, Br) and Bi2MoO6 etc., have aroused increasing interest due to the unique electron structure of Bi element.9,10 Among them, Bi2MoO6 (band gap ∼ 2.7 eV) is a simple n-type semiconductor with a layered Aurivillius structure, which has been reported as a promising candidate of visible-light-responsive photocatalysts.11,12 However, the overall efficiency of single phase Bi2MoO6 is still low, due to the rapid recombination of electron–hole pairs after excitation. To better improve the visible-light-responsive photocatalytic activity, a variety of strategies like rare earth elements doping, morphology controlled synthesis and heterojunctions construction have been developed to improve the performance.13–17 Semiconductor heterojunction presents great potential application because of their tunable light absorption and the effective inhibition of the recombination of photogenerated carries.18,19 heterostructured Bi2O2CO3/Bi2MoO6 nanocomposites were reported with higher activity and stability for the photocatalytic reactions. A high-efficient gradient charge transfer was observed in the Bi2MoO6/Bi2WO6 heterojunction for the decolorization of methylene blue (MB).20 C60, C3N4 and carbon nanofibers modified Bi2MoO6 showed the superior photocatalytic properties than that of the pure component under the visible light excitation. α-Bi2O3 is a p-type semiconductor with suitable band gap potential (∼2.8 eV) for photocatalytic reactions.21 Xu et al. reported that Bi2O3/Bi2MoO6 microspheres synthesized via the solvothermal method showed good properties of antimicrobial effect and the decomposition of RhB under visible light excitation.22 Combining the p-type Bi2O3 together with the n-type Bi2MoO6 can greatly inhibited the recombination of photoinduced carriers by the formation of inner electric field in the p–n junction. In composite photocatalysts, the component interfaces make very important contribution to the enhancement of photodegradation activity.23
In this work, we reported the synthesis of Bi2MoO6/Bi2O3 heterojunctions through an in situ alkali etching growth method, which is a low-cost and easily controlled way to obtain the good connection of the heterojunction interfaces. The photocatalytic activity of Bi2MoO6/Bi2O3 samples was evaluated by photodecomposition of RhB and the photocatalytic mechanism was discussed based on the scavengers studies.
2. Experimental section
2.1 Preparation of the Bi2MoO6
Bi2MoO6 nanoplates were synthesized through a hydrothermal process using commercial chemicals of analytical grade without further purification. Briefly, Bi(NO3)3·5H2O (0.8 g) and (NH4)6Mo7O2·4H2O (0.15 g) were completely dissolved in 12 mL ethanol and 10 mL H2O, respectively. After stirring for 30 min, both of the ethanol and H2O solutions were transferred into a 30 mL Teflon-lined stainless autoclave with an addition of 2 mL 0.8 M NaOH solution. The autoclave was heated at 150 °C for 22 h in an oven. Then, the reactor was cooled down to room temperature. The resulting powder with yellow-green colour was filtered and washed several times with deionized water and finally dried in air at 60 °C for 6 h.
2.2 The alkali etching of Bi2MoO6
The etching process was carried out by adding 0.1 g Bi2MoO6 powders in 30 mL 0.1 M NaOH solution for a certain time. And then, the resulting powder was filtered and washed with deionized water for several times. The final products were dried in air at 60 °C for 6 h. All the Bi2MoO6/Bi2O3 composites were prepared in such an etching way with different reaction times, which were labeled as E15, E30, E45 and E60 for the composites with etching duration of 15 min, 30 min, 45 min and 60 min, respectively.
2.3 Characterization
The crystal structure of Bi2MoO6 nanoplate and Bi2MoO6/Bi2O3 composites was analyzed by the Rigaku D/max-2600/PC X-ray diffractometer using Cu-Kα radiation (λ = 1.54178 Å) over the 2θ range of 10–70°. Transmission electron microscopy (TEM) analysis was measured by a FEI Tecai G2 F20 at a 200 kV accelerating voltage. The morphology change of the Bi2MoO6/Bi2O3 photocatalysts was characterized by a scanning electron microscopy (SEM, Hitachi SU-70). Elementary composition was examined by X-ray photoelectron spectroscopy (XPS, Thermofisher K-Alpha) with Al source. The standard binding energy of C1s is 285 eV. The optical properties of the composites were determined by the UV-vis diffuse reflectance spectra (Cary 500 UV-vis-NIR spectrophotometer) over the wavelength range of 200 nm to 700 nm. Specific surface areas were determined from nitrogen adsorption–desorption isotherms using a surface area analyzer (Quantachrome NOVA 2000e) and calculated by the Brunauer–Emmett–Teller (BET) method.
2.4 Photocatalytic activity and photocurrent spectra measurements
The photocatalytic activity test was carried out by irradiation RhB-photocatalysts suspensions under a 500 W xenon lamp (cutoff filter λ > 400 nm). The molecular structure of RhB was shown in Fig. S1.† In each test, 0.02 g photocatalysts were dispersed into the RhB dye solution (1 × 10−5 mol L−1 60 mL) and stirred for 40 min in the darkness in order to achieve the desorption–adsorption equilibrium of dye on the surface of catalysts. After the desorption–adsorption process, the light source irradiated on the suspension with a distance of 10 cm. During the photocatalytic reaction, 4 mL liquid was taken from the reactor at a time interval of 20 min followed by the separation of photocatalyst through centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm min−1 for 5 min). The concentration of RhB solution without catalyst was determined at 554 nm using a spectrophotometer (Perkin-Elmer Lambda-35 UV-vis spectrometer). The total organic carbon (TOC) concentration was measured using a Multi TOC/TN Analyzer NC 2100 s (Analytik Jena AG Corporation).
000 rpm min−1 for 5 min). The concentration of RhB solution without catalyst was determined at 554 nm using a spectrophotometer (Perkin-Elmer Lambda-35 UV-vis spectrometer). The total organic carbon (TOC) concentration was measured using a Multi TOC/TN Analyzer NC 2100 s (Analytik Jena AG Corporation).
The photocurrent spectra of pure and E45 sample were measured by an electrochemical workstation (CHI660E, Chenhua Instruments) with a standard three-electrode configuration. Fluorine-doped tin oxide (FTO) loaded with catalyst, Pt wire and saturated calomel electrode were employed as working electrode, counter electrode and reference electrode, respectively. Typically, 10 mg of sample powder was dispersed into 2 mL of N,N-dimethylformamide under ultrasonication for 10 min to obtain slurry, which was further spread onto FTO glasses with conductive glue to obtain sample films with an area of 1 cm × 1 cm. As-prepared FTO glass electrodes were dried at 100 °C for 60 min under ambient conditions to improve adhesion. 50 mL of 0.5 M Na2SO4 (pH = 6.8) was used as the electrolyte solution. A 300 W xenon lamp with a 420 nm cut-off filter was employed as a visible light photosource.
3. Results and discussion
3.1 Structure and morphology
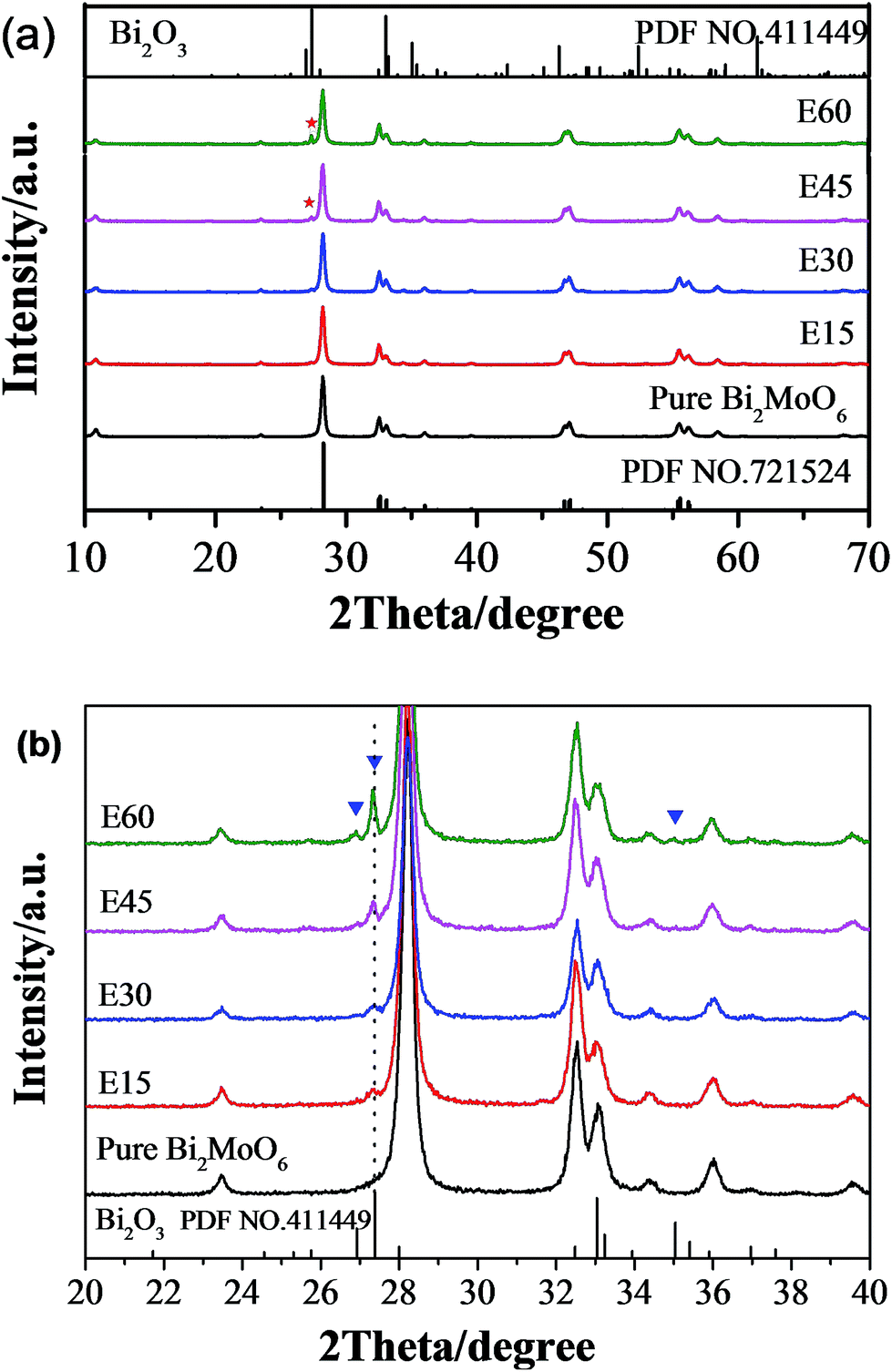
The XRD patterns of fresh Bi2MoO6 and Bi2MoO6/Bi2O3 composites are shown in Fig. 1. The profile in Fig. 1(a) confirms that as-prepared powder is phase-pure orthorhombic Bi2MoO6, which is consistent with the reported result (JCPDS cards no. 72-1524). After alkaline etching process, small peaks at around 27° and 35° become detectable, which was ascribed to the formation of monoclinic α-Bi2O3 phase (JCPDS cards no. 41-1449). As the duration of etching process prolonged, new peaks turn to be stronger, especially in E45 and E60 samples, indicating the amount of α-Bi2O3 phase increases with the prolonged etching treatment. The formation of α-Bi2O3 is different from the work by Han et al., who reported that γ-Bi2O3 nanoparticles were formed on m-BiVO4 surface by NaOH alkaline “etching”.24 This could be due to the different reaction conditions for the etching treatment. The etching of m-BiVO4 was carried out at a high temperature and high pressure condition (130 °C in an autoclave), while, in our study, it was performed at room temperature. Considering the intensity of diffraction peaks is proportional to the quantity of crystalline phase, it can be deduced that the composite is composed of Bi2MoO6 host and small amount of α-Bi2O3.25 Moreover, all the diffraction peaks of E15, E30, E45 and E60 are sharp and strong, indicating the good crystallinity of the composites.
 |
| | Fig. 1 XRD patterns of Bi2MoO6 composites with variable etching time. Referenced standard positions of diffraction peaks were taken from the JCPDS card no. 72-1524 for Bi2MoO6 and JCPDS card no. 41-1449 for Bi2O3 as denoted. (b) an enlarge view of XRD patterns in the 2θ range of 20–40°. | |
The morphology of samples was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As can be seen in Fig. 2(a), the as-synthesized pure Bi2MoO6 is composed of many nanosheets. After the etching process treatment, a morphology change in etched Bi2MoO6 samples can be observed. A typical SEM image of sample E45 nanosheet in Fig. 2(b) reveals that the edges of nanosheets turn to be round-like and fuzzy. A TEM image in Fig. 2(c) of E45 sample also illustrates its nanosheets morphology, in good agreement in the SEM observation. Further, the HRTEM image (Fig. 2(d)) from the selected region of Fig. 2(c) shows that the spacings of the lattice fringes were found to be 0.276 nm, corresponding to distances of the (200) of orthorhombic Bi2MoO6.26,27 While the lattice fringe distance of 0.331 nm agrees well with the (111) lattice planes of α-Bi2O3, further confirming the formation of α-Bi2O3 phase after the etching process. From the XRD and TEM analysis, it can be considered that the alkali etching of Bi2MoO6 is favorable for the formation of Bi2O3 on Bi2MoO6 surface. The good connection between the α-Bi2O3 nanocrystals and the Bi2MoO6 host provides a smooth pathway for the photoinduced electrons transfer, reducing the recombination probability of photocarriers.
 |
| | Fig. 2 SEM images of (a) pure Bi2MoO6 (b) E45, TEM image of (c) E45 and (d) HRTEM image of the selected region from E45. | |
Based on the analysis given above, the formation of α-Bi2O3 phase on Bi2MoO6 surface via alkaline etching can be described in Scheme 1. It is well-known that Aurivillius Bi2MoO6 is built up of alternating perovskite-like (MoO4)2− and fluorite-like (Bi2O2)2+ layers.28 In alkaline condition, the outer surface of Bi2MoO6 loses molybdenum. But Bi3+ ions are not stable in alkaline solution and these Bi species can precipitate on the outer surface of Bi2MoO6 host and reconstruct as α-Bi2O3 phase. With the increasing of etching time, more molybdenum dissolved, leading to increased amount of α-Bi2O3 nanoparticles. After etching treatment, the single phase Bi2MoO6 crystals were transformed into the Bi2MoO6/α-Bi2O3 heterostructure, which is beneficial for the improvement of photocatalytic activity of Bi2MoO6.
 |
| | Scheme 1 Schematic alkali etching process and the formation of Bi2MoO6/Bi2O3 composite. | |
3.2 XPS analysis
In order to obtain detailed information regarding to the chemical and bonding environment of elements, samples of pure and E45 were examined by X-ray photoelectron spectroscopy. The representative XPS surveys were shown in Fig. 3(a) in the range of 0–1200 eV, which clearly demonstrate the existence of Bi, O and Mo elements. C1s peak can be ascribed to the adventitious hydrocarbon from XPS instrument. Both Bi4f and Mo3d show two peaks, as depicted in the high-resolution spectra in Fig. 3(b and d), due to the spin–orbit split effect. Two strong Bi peaks centered at 164.5 and 159.1 eV are associated with core lines of Bi4f5/2, and 4f7/2, respectively. After the etching for 45 minutes, Bi states in E45 sample can be divided into four peaks. The values of 164.6 and 159.3 eV can be ascribed to the Bi3+ of the Bi2MoO6, while the values of 164.2 eV and 158.6 eV belong to the Bi3+ in the Bi2O3, which further confirms the existence of Bi2O3 in the composite.29 The BE values of Mo3d centered at 232.2 eV and 235.3 eV (Fig. 3(c)) is in agreement with the literature.30 In Fig. 3(d), the O can be fitted into three kinds of chemical states, indicating that at least three kinds of oxygen species are presented in the near-surface region. The binding energies at 529.7 eV and 530.2 eV can be assigned to the crystal lattice oxygen of Bi2MoO6 and Bi2O3, respectively, while the peak at 530.8 eV can be assigned to the adsorbed oxygen, including hydroxyl and carbonate groups adsorbed on the material surface.31,32 From the XPS analysis, we can also confirm that the etching process leads to the formation of Bi2O3 in the near-surface region, in accordance with the XRD and TEM analysis.
 |
| | Fig. 3 XPS spectra of the pure Bi2MoO6 and E45 (a) survey spectrum (b) Bi4f (c) Mo3d and (d) O1s. | |
3.3 Optical absorption property
The optical properties of the Bi2MoO6/Bi2O3 composites were characterized through the UV-vis absorption spectra and the results are shown in Fig. 4(a). All the Bi2MoO6/Bi2O3 composite materials exhibit photo-absorption from the UV light region to the visible-light region with the wavelength shorter than 460 nm. Compared with the pure Bi2MoO6, the composites present a slightly increase in the light absorption. The steep shape of the spectra indicated that the visible-light absorption is arisen from the band-gap transition instead of the impurity level.16 From the absorption spectra, the band gap energy (Eg) of the samples can be determined by the formula of αhν = A(hν − Eg)n/2, here, α, h, ν, Eg and A are absorption coefficients, Planck constant, light frequency, band gap and a constant, respectively.33 The Eg values of the samples pure, E15, E30, E45, E60 can be estimated as 2.68, 2.62, 2.69, 2.71, 2.66 eV, respectively. For the pure Bi2MoO6, the Eg value is consistent with the reported literature.34 With the increase of etching duration, it can be found that the band gap is generally increased from 2.68 eV to 2.71 eV, due to the increased amount of α-Bi2O3 (∼2.8 eV) on the Bi2MoO6 surface.35 Similar phenomenon has been observed in BiVO4@Bi2O3 core–shell microspheres, which show a larger band-gap energy (2.52 eV) than single-phase BiVO4 (2.43 eV).36
 |
| | Fig. 4 (a) UV-visible diffuse reflectance spectra of the pure Bi2MoO6 and Bi2MoO6/Bi2O3 composite materials with different reaction time, (b) the estimated band gap energies of Bi2MoO6/Bi2O3 composite samples. | |
3.4 Photocatalytic activity and the recycling test
The photocatalytic activity was evaluated by the decomposition of RhB solution under visible light excitation as a function of time (Fig. 5(a)). Further information about RhB was provided in electronic ESI (Fig. S1†). Pure Bi2MoO6 and the etched samples were all examined for comparison under the same photocatalytic experimental condition. Pure Bi2MoO6 presents weak photocatalytic activity under visible light irradiation. After the etching treatment for a certain time, an enhanced photocatalytic activity can be found. The highest photocatalytic activity is obtained on E45 sample, which decomposed about 98% of RhB dye molecule after the irradiation for 100 min. In comparison, the degradation efficiency of pure, E15, E30 and E60 only reach about 18%, 47%, 73% and 47%, respectively.
 |
| | Fig. 5 (a) Visible-light photocatalytic degradation of RhB solution over the products prepared at different etching time. (b) Stability of the sample E45 on the photocatalytic degradation of RhB for reusing 5 times. | |
The photodegradation kinetics was studied through plotting the ln(C/C0) as a function of reaction time which exhibits linear relationships. The pseudo-first order rate constants (k min−1) were determined from the slope of the lines and the apparent kinetic constants k for the photodegradation of RhB as a function of the etching time was plotted (Fig. S2†). It shows that the kinetic constant increases with the increasing of etching time, and the highest value is obtained at E45 sample with an etching time of 45 min. The apparent rate constant for E45 is 5.1 times higher than that of pure sample, implying the high photocatalytic performance.
Note that all the etched samples the exhibit better photocatalytic performance than the pure one, which can be attributed to the inner electric field formed by the p–n junction interfaces in the composites. These interfaces can effectively separate the photocarries irradiated by the visible light. Moreover, since the Bi2O3 crystalline formed on the Bi2MoO6 surface are in nanosize, the photoinduced electrons from Bi2O3 can easily move to the surface for the photocatalytic reaction or transfer across the interface of p–n junction to the band gap of Bi2MoO6. On the other hand, a declining is observed with the etching time increasing to 60 min, probably due to the much heavier content of Bi2O3 loading. The content of Bi2O3 nanocrystals elevates as the etching process prolonged and more surface of Bi2MoO6 is covered by Bi2O3, which has a higher band gap (2.8 eV) than that of Bi2MoO6 (2.67 eV). The surplus of Bi2O3 nanocrystals could inhibit the light absorption, increase the possibility of the photocarries recombination and also reduce the reaction sites on the surface of Bi2MoO6, leading to the decrease of photocatalytic activity. Anyway, our experimental results show that the proper etching synthesis of Bi2MoO6/Bi2O3 heterojunction is a feasible and controllable strategy to enhance the photocatalytic activity of phase-pure Bi2MoO6.
For practical photocatalytic application like waste water purification, the recycling of photocatalysts is an important parameter for the industrial application of photocatalysts. To test the reusability of the Bi2MoO6/Bi2O3 photocatalysts, the E45 sample was used for five cycles under the same condition, and the result is shown in Fig. 5(b). After five catalytic runs, the catalyst also remains superior activity with 93% degradation rate, which confirms that Bi2MoO6/Bi2O3 heterojunction has high stability and is easily to be used for recycling.
3.5 Photocatalytic activity mechanism
α-Bi2O3 is a p-type semiconductor, whereas Bi2MoO6 is a n-type semiconductor. When Bi2MoO6 combines with α-Bi2O3, a p–n junction will form and the charge carries will diffuse in opposite direction to form an internal electric field at the heterojunction interface.22 The CB edge potential of Bi2MoO6 (−0.32 eV) is more negative than α-Bi2O3 (0.33 eV).25,37 Thus, under the thermal equilibrium conditions, the Fermi levels of Bi2MoO6 and α-Bi2O3 are re-aligned; the energy band positions of Bi2MoO6 shifts to the downward direction and that of α-Bi2O3 shifts toward the upward direction, as shown in Scheme 2. The internal electric field forms by the p–n junction directed from the n-type Bi2MoO6 to the p-type Bi2O3 is simultaneously built along with the Fermi level alignment. Therefore, the band positions of the p-type α-Bi2O3 and n-type Bi2MoO6 in the heterojunction present a type-II band structure (Scheme 2).38 After the visible-light excitation, both Bi2MoO6 and α-Bi2O3 could be easily excited and corresponding photoinduced electron–hole pairs are generated as described in eqn (1). The photoexcited electrons excited from the valance band of α-Bi2O3 will be transferred to the conduction band of Bi2MoO6. Meanwhile, the photo-excited holes on the valence band of Bi2MoO6 will prefer to flow down to the valence band of α-Bi2O3 crossing the interfaces (eqn (2)). Such transportation of the photogenerated carriers could extend their transfer path or stabilize the photogenerated holes in the valance band of α-Bi2O3, leading to the prolonged lifetime of the charge carriers and successfully hindering the unfavorable recombination of electron–hole pairs.39 BET specific surface areas were calculated from the N2 adsorption–desorption isotherms (Fig. S3†) for the pure Bi2MoO6 (16.82 m2 g−1) and E45 (17.72 m2 g−1). The specific surface area of Bi2MoO6 is slightly increased after etching treatment, which is considered to play a less important role for significant activity improvement. This result further confirms that the performance enhancement is mainly induced by the effective separation between electron and holes from the heterostructure interfaces.
 |
| | Scheme 2 Schematic illustration of the charge transfer pathway during the RhB degradation process over Bi2MoO6/Bi2O3 under visible light irradiation. | |
Zou et al. have reported that the photodegradation of RhB is dominated by the photooxidation process.40 Thus, it is necessary to identify the main oxidant in decolorizing RhB over the Bi2MoO6/Bi2O3 heterojunctions. Generally, introducing scavengers of holes and hydroxyl radicals into the photocatalytic reaction is an effective way to identify the main active species.41 In this study, ethylenediaminetetraacetate (EDTA) and tert butyl alcohol (TBA) were respectively used as holes and hydroxyl radicals scavengers and their effects on the photocatalytic degradation on RhB is shown in Fig. 6. After the addition of TBA (hydroxyl radicals quencher), the degrading rate of RhB was obviously depressed, indicating that hydroxyl radicals are the active species in the reaction. Considering the generation of hydroxyl radicals, two possible ways would be promoted.40 One way is the direct oxidation products of holes. Generally, the energy of photogenerated carrier (holes and electrons) is approximately equal to the band energy (valence band and conduction band).1 It has been reported that the valence band of Bi2MoO6 is about 2.44 V vs. NHE, which means that the energy of photoinduced holes are inefficient to directly oxidize adsorbed hydroxyl groups for generating hydroxyl radicals (2.7 V vs. NHE).40,42 The other one is a multistep reduction of O2 dissolved in the solution. Based on the band gap analysis in this work, the energy of electrons from the conduction band of Bi2MoO6 (−0.23 V vs. NHE) is sufficient to produce hydroxyl radicals via the reduction of dissolved O2 (+0.13 V).42 Thus, hydroxyl radicals were one of the important active species in the degradation of RhB over Bi2MoO6/Bi2O3 heterojunctions (eqn (3)). In the case of EDTA (h+ quencher) addition, it drastically quenched the photodecomposition rate of RhB, which indicates that the dominate active species are h+. The redox potential of RhB is 1.43 V,43 which is much lower than that of h+ coming from the valance band of Bi2MoO6. It is reasonable to consider that the degradation of RhB could be conducted by the direct oxidation of photoinduced holes from the Bi2MoO6 (eqn (4)).
| | |
(Bi2MoO6/Bi2O3) + hv → (Bi2MoO6/Bi2O3)(eCB− + hVB+)
| (1) |
| | |
Bi2MoO6(eCB−⋯hVB+)/Bi2O3(eCB−⋯hVB+) → Bi2MoO6·eCB−/Bi2O3·hVB+
| (2) |
| | |
˙OH + RhB → intermediate products → CO2+H2O
| (3) |
| | |
hVB+ + RhB → intermediate products → CO2 + H2O
| (4) |
 |
| | Fig. 6 Effects of EDTA and TBA on the decomposition of RhB in the presence of E45. | |
The measurement of photocurrent was carried out to study the efficiency of the separation of photogenerated electron–hole pairs within the photoelectrode.44 Fig. 7 shows the photocurrents of the pure and E45 with the light irradiation off and on. An obvious increase of the photocurrent intensity in E45 is observed compared to the pure sample. The high current density indicates that the photogenerated electrons and holes of etched Bi2MoO6 prefer to separate and transfer to the electrode due to the heterostructures formed between Bi2MoO6 and Bi2O3. Thus, the etching process is a facile way to obtain an effective interface for the separation of photoinduced carriers and promoting the photocatalytic activities.
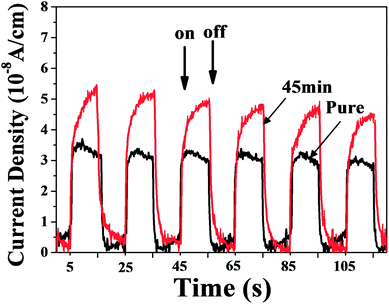 |
| | Fig. 7 Photocurrent responses of the pure and E45 under visible light irradiation. | |
Generally, the mineralization of the organic dye can also lead to the decolorization, which does not mean obtaining the final products such as H2O and CO2. Therefore, it is necessary to test the TOC degradation of dyes. As can be seen from the experimental results (Fig. 8), the decolorization process is more beneficial than TOC removal. When the decolorization is about 98%, the TOC removal was estimated to be 46.5%. The undecomposed organic dye can be observed by the UV-vis spectral changes of RhB. The inserted figure shows that the main absorbance of 554 nm shifted gradually to the shorter wavelength 498 nm, which suggests that the stepwise formation of a series of N-deethylated intermediates as the irradiation time prolonged.45 This further indicates that the RhB decolorization is partially decomposed into H2O and CO2.
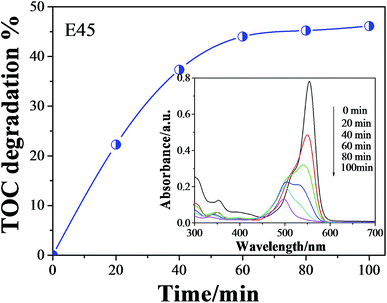 |
| | Fig. 8 TOC degradation as a function of time for the decomposition of RhB using E45 as photocatalyst. Inserted: UV-vis spectral changes of RhB (1.0 × 10−5 M) in aqueous E45 dispersions as a function of irradiation time. | |
4. Conclusion
In summary, heterojunction Bi2MoO6/Bi2O3 composites were successfully prepared and evaluated as effective visible-light-responsive photocatalysts. From the results and discussion above, we can come to the conclusions as follows: (1) Bi2MoO6/Bi2O3 heterojunctions with different etching time were prepared via a facile alkali etching method and XRD, TEM and XPS analysis confirmed the formation of α-Bi2O3 nanophase on Bi2MoO6 surface. (2) Bi2MoO6/Bi2O3 heterojunctions exhibited improved visible-light activity than pure Bi2MoO6, and E45 sample exhibited highest performance with nearly 98% degradation of RhB after 100 min irradiation. (3) The high photodecomposition activity can be attributed to the good visible light absorption of both Bi2MoO6 and Bi2O3 as well as the efficient separation of photoinduced electron–hole pairs through the Bi2MoO6/Bi2O3 heterojunction interface.
Acknowledgements
This work was supported by the National Science Foundation of China (51102069), the Outstanding Youth Science Foundation of Harbin Normal University, Innovative Talents Fund of Harbin (2014RFQXJ085) and Open Project from Key Laboratory for Photonic and Electronic Bandgap Materials (PEBM 201209). The authors are greatly thankful for the valuable discussion with Dr Wenjuan Yang.
References
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- Z. G. Yi, J. H. Ye, N. Kikugawa, T. Kako, S. X. Ouyang, H. Stuart-Williams, H. Yang, J. Y. Cao, W. J. Luo, Z. S. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- S. Zheng, W. Jiang, Y. Cai, D. D. Dionysiou and K. E. O'Shea, Catal. Today, 2014, 224, 83–88 CrossRef CAS PubMed.
- W. J. Jiang, J. A. Joens, D. D. Dionysiou and K. E. O'Shea, J. Photochem. Photobiol., A, 2013, 262, 7–13 CrossRef CAS PubMed.
- ACS Symposium Series, ed. N. Shamim and V. K. Sharma, American Chemical Society, Washington, DC, USA, 2013, ch. 12, pp. 201–229 Search PubMed.
- W. D. Zhang and L. Zhu, J. Nanosci. Nanotechnol., 2012, 12, 6294–6300 CrossRef CAS PubMed.
- G. H. Tian, Y. J. Chen, R. T. Zhai, J. Zhou, W. Zhou, R. H. Wang, K. Pan, C. G. Tian and H. G. Fu, J. Mater. Chem. A, 2013, 1, 6961–6968 CAS.
- M. Y. Wang, J. Ioccozia, L. Sun, C. J. Lin and Z. Q. Lin, Energy Environ. Sci., 2014, 7, 2182–2202 CAS.
- W. Z. Wang, M. Shang, W. Z. Yin, J. Ren and L. Zhou, J. Inorg. Mater., 2012, 27, 11–18 CrossRef CAS.
- F. Duan, Q. Zhang, Q. F. Wei, D. J. Shi and M. Q. Chen, Prog. Chem., 2014, 26, 30–40 Search PubMed.
- Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2006, 110, 17790–17797 CrossRef CAS PubMed.
- S. C. Zhang, C. A. Zhang, Y. Man and Y. F. Zhu, J. Solid State Chem., 2006, 179, 62–69 CrossRef CAS PubMed.
- A. A. Alemi, R. Kashfi and B. Shabani, J. Mol. Catal. A: Chem., 2014, 392, 290–298 CrossRef CAS PubMed.
- Y. C. Miao, G. F. Pan, Y. N. Huo and H. X. Li, Chin. J. Inorg. Chem., 2014, 30, 1587–1592 CAS.
- A. Phuruangrat, N. Ekthammathat, B. Kuntalue, P. Dumrongrojthanath, S. Thongtem and T. Thongtem, J. Nanomater., 2014, 2014, 934165 Search PubMed.
- L. W. Zhang, T. G. Xu, X. Zhao and Y. F. Zhu, Appl. Catal., B, 2010, 98, 138–146 CrossRef CAS PubMed.
- J. H. Bi, J. G. Che, L. Wu and M. H. Liu, Mater. Res. Bull., 2013, 48, 2071–2075 CrossRef CAS PubMed.
- Y. S. Xu and W. D. Zhang, Appl. Catal., B, 2013, 140, 306–316 CrossRef PubMed.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- F. J. Zhang, S. F. Zhu, F. Z. Xie, J. Zhang and Z. D. Meng, Sep. Purif. Technol., 2013, 113, 1–8 CrossRef CAS PubMed.
- J. G. Hou, C. Yang, Z. Wang, W. L. Zhou, S. Q. Jiao and H. M. Zhu, Appl. Catal., B, 2013, 142, 504–511 CrossRef PubMed.
- Y. S. Xu, Z. J. Zhang and W. D. Zhang, Mater. Res. Bull., 2013, 48, 1420–1427 CrossRef CAS PubMed.
- Y. Sasaki, A. Tanaka, K. Hashimoto and H. Kominami, Chem. Lett., 2013, 42, 419–421 CrossRef CAS.
- M. D. Han, T. Sun, P. Y. Tan, X. F. Chen, O. K. Tan and M. S. Tse, RSC Adv., 2013, 3, 24964–24970 RSC.
- M. Y. Zhang, C. L. Shao, J. B. Mu, Z. Y. Zhang, Z. C. Guo, P. Zhang and Y. C. Liu, CrystEngComm, 2012, 14, 605–612 RSC.
- H. P. Li, J. Y. Liu, W. G. Hou, N. Du, R. J. Zhang and X. T. Tao, Appl. Catal., B, 2014, 160, 89–97 CrossRef PubMed.
- J. L. Long, S. C. Wang, H. J. Chang, B. Z. Zhao, B. T. Liu, Y. G. Zhou, W. Wei, X. X. Wang, L. Huang and W. Huang, Small, 2014, 10, 2791–2795 CrossRef CAS PubMed.
- G. Sankar, M. A. Roberts, J. M. Thomas, G. U. Kulkarni, N. Rangavittal and C. N. R. Rao, J. Solid State Chem., 1995, 119, 210–215 CrossRef CAS.
- J. H. Bi, L. Wu, H. Li, Z. H. Li, X. X. Wang and X. Z. Fu, Acta Mater., 2007, 55, 4699–4705 CrossRef CAS PubMed.
- M. Y. Zhang, C. L. Shao, J. B. Mu, X. M. Huang, Z. Y. Zhang, Z. C. Guo, P. Zhang and Y. C. Liu, J. Mater. Chem., 2012, 22, 577–584 RSC.
- J. H. Bi, J. Li, Z. H. Li, X. X. Wang and X. Z. Fu, Acta Mater., 2007, 55, 4699–4705 CrossRef CAS PubMed.
- L. Zhang, Y. Man and Y. Zhu, ACS Catal., 2011, 1, 841–848 CrossRef CAS.
- J. Zhang, F. J. Shi, J. Lin, D. F. Chen, J. M. Gao, Z. X. Huang, X. X. Ding and C. C. Tang, Chem. Mater., 2008, 20, 2937–2941 CrossRef CAS.
- X. Zhao, J. H. Qu, H. J. Liu and C. Hu, Environ. Sci. Technol., 2007, 41, 6802–6807 CrossRef CAS.
- L. Zhu, B. Wei, L. L. Xu, Z. Lu, H. L. Zhang, H. Gao and J. X. Che, CrystEngComm, 2012, 14, 5705–5709 RSC.
- M. L. Guan, D. K. Ma, S. W. Hu, Y. J. Chen and S. M. Huang, Inorg. Chem., 2011, 50, 800–805 CrossRef CAS PubMed.
- M. S. Gui, W. D. Zhang, Q. X. Su and C. H. Chen, J. Solid State Chem., 2011, 184, 1977–1982 CrossRef CAS PubMed.
- Y. J. Wang, Q. S. Wang, X. Y. Zhan, F. M. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- L. Kong, Z. Jiang, H. H. Lai, R. J. Nicholls, T. C. Xiao, M. O. Jones and P. P. Edwards, J. Catal., 2012, 293, 116–125 CrossRef CAS PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- F. Zhou, R. Shi and Y. F. Zhu, J. Mol. Catal. A: Chem., 2011, 340, 77–82 CrossRef CAS PubMed.
- Y. S. Xu and W. D. Zhang, Dalton Trans., 2013, 42, 1094–1101 RSC.
- T. Shen, Z. G. Zhao, Q. Yu and H. J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203–212 CrossRef CAS.
- A. Hagfeldt, H. Lindstrom, S. Sodergren and S. E. Lindquist, J. Electroanal. Chem., 1995, 381, 39–46 CrossRef.
- T. Jia, W. M. Wang, F. Long, Z. Y. Fu, H. Wang and Q. J. Zhang, J. Phys. Chem. C, 2009, 113, 9071–9077 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14184a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.