
The efficient and selective biocatalytic oxidation of norisoprenoid and aromatic substrates by CYP101B1 from Novosphingobium aromaticivorans DSM12444†
Emma A. Hall and
Stephen G. Bell*
School of Chemistry and Physics, University of Adelaide, SA 5005, Australia. E-mail: stephen.bell@adelaide.edu.au; Fax: +61 8 83034380; Tel: +61 8 83134822
First published on 12th December 2014
Abstract
CYP101B1 from Novosphingobium aromaticivorans DSM12444 is a homologue of CYP101A1 (P450cam) from Pseudomonas putida and the CYP101D1, CYP101D2 and CYP101C1 enzymes from the same bacterium. CYP101B1 binds norisoprenoids more tightly than camphor and efficiently hydroxylates substrates in combination with ferredoxin reductase, ArR, and [2Fe–2S] ferredoxin, Arx, electron transfer partners. The norisoprenoids, α-ionone and β-damascone are both oxidised by CYP101B1 with high product formation activity, >500 min−1. α-Ionone oxidation occurred regioselectively at the allylic C3 position while β-damascone was hydroxylated predominantly at C3, 86%, with the main competing minor product arising from oxidation at the allylic C4 position (11%). When incorporated into a whole-cell oxidation system, with ArR and Arx, CYP101B1 is also capable of oxidising the aromatic compound indole. Other aromatic molecules including phenylcyclohexane and p-cymene were tested and both were hydroxylated by CYP101B1. Phenylcyclohexane was selectively oxidised to trans-4-phenylcyclohexanol while p-cymene was hydroxylated at the benzylic carbons to yield a mixture of isopropylbenzyl alcohol and p-α,α-trimethylbenzylalcohol. Trans-4-Phenylcyclohexanol was formed with a product formation rate of 141 min−1 and was five times more active than the oxidation of p-cymene.
Introduction
Cytochrome P450 (CYP) enzymes constitute a superfamily of heme-containing monooxygenases which, among other activities, are able to insert one oxygen atom from dioxygen into chemically inert carbon–hydrogen bonds with high regio- and stereoselectivity.1,2 They are of great interest due to their myriad of physiological roles and potential application in the biocatalytic synthesis of fine chemicals under mild conditions.3–5 Dioxygen activation requires two electrons that are usually derived from NAD(P)H and delivered to the CYP enzymes by electron transfer proteins.6 Numerous CYP enzymes with potentially interesting and desirable activities have been discovered but many of these are orphaned, with no electron transfer partners located nearby in the genome, and their activities are often compromised in hybrid electron transfer systems.7,8 Therefore, the identification and characterisation of CYP enzymes with functional and highly active electron transfer chains is imperative in order to optimise the catalytic activity of this class of enzymes across a broad range of substrates and reaction types.We have reported the heterologous production and purification of the majority of the sixteen P450 enzymes from Novosphingobium aromaticivorans DSM12444.5,9 Potential substrates for many of these enzymes have been identified and these span a broad range of organic molecules including terpenoids, linear alkanes and polyaromatic hydrocarbons.5,10,11 The CYP enzymes of this bacterium are presumably involved in metabolic processes which enable it to survive in the oligotrophic environments where it lives.12,13 A class I electron transfer system, consisting of a flavin-dependent ferredoxin reductase, ArR, and a [2Fe–2S] ferredoxin, Arx, has been identified which is able to reconstitute the monooxygenase activity of at least five of these enzymes (CYP101B1, CYP101C1, CYP101D1, CYP101D2 and CYP111A2).5,9,14 Whole-cell systems capable of product formation on the gram-per-litre scale in shake flasks have also been constructed.9 Like P450cam (CYP101A1), CYP101D1 and CYP101D2 both bind and oxidise camphor (yielding ≥98% 5-exo-hydroxycamphor). While CYP101B1 catalyses the unselective oxidation of camphor generating four products, CYP101C1 does not. CYP101C1 and CYP101B1 are able to bind and oxidise β-ionone. Crystal structures of CYP101D1, CYP101D2 and CYP101C1 and the ArR and Arx electron transfer partners reveal striking similarities between these CYP101 family enzymes and CYP101A1.15–18 The enzymes also provide templates in order to understand how variations in the amino acid sequence of closely related P450 enzymes can lead to different substrate binding preferences, altered product selectivity and a predilection for specific electron transfer partners.15–17
CYP101B1 when combined with ArR and Arx in a whole-cell oxidation system was found to turn the culture medium blue indicating that it is able to hydroxylate indole to 3-hydroxyindole which can then dimerise to form indigo.9,19 Novosphingobium species are able to degrade a wide variety of aromatic hydrocarbons and therefore CYP101B1 has the potential to be a biocatalyst for the oxidation of these hydrophobic substrates.9,12,13 Here we report our investigations into the substrate range of CYP101B1 and show it is a highly active and regioselective biocatalyst for the oxidation of α-ionone and β-damascone. We also investigate the ability of CYP101B1 to oxidise hydrophobic aromatic ring containing substrates such as phenylcyclohexane and p-cymene.
Results
Phylogenetic analysis of CYP101B1
CYP101B1 shares 46% sequence identity with CYP101D2 and 44% with CYP101A1, and CYP101D1 (Fig. 1 and Table S1†). A BLAST search revealed only one CYP enzyme in the NCBI database, from Novosphingobium tardaugens NBRC 1675, with a greater than 50% identity to CYP101B1 (59%). Other CYP enzymes identified in the 45–50% range all have greater sequence homology with CYP101A1 or CYP101D2. As a result there are currently only two identified members of the CYP101B subfamily and both are found in species of Novosphingobium bacteria. CYP101B1 has little sequence similarity with its closest structurally characterised relative outside of the CYP101 family, CYP176A1 (P450cin) or other ionone binding P450 monooxygenases such as CYP109D1 and CYP264B1, or those from Streptomyces species which are known to oxidise norisoprenoids (Table S1,† vide infra).20–23 | ||
| Fig. 1 Protein sequence alignments of CYP101B1, CYP101D1, CYP101D2 and CYP101C1 from N. aromaticivorans, CYP101A1 (P450cam) from a Pseudomonas putida species and CYP101B2 from Novosphingobium tardaugens NBRC 16725. Conserved residues are highlighted in red boxes with similar residues in red font. | ||
Despite the relatively low sequence similarity with CYP101B2 from N. tardaugens a comparison of the residues that are likely to make up the active site and substrate access channel revealed the almost complete conservation of amino acids (Fig. 1 and Table S2†). The only difference was the conservative replacement of Ile236 in CYP101B1 with Val244 in CYP101B2. This is the equivalent position to Val247 in CYP101A1 a residue located in the active site which is important for substrate binding.24 This comparison also revealed important differences in the active sites of the two CYP101B subfamily enzymes when compared to the other members of the CYP101 family. For example sequence alignment reveals that, Val91 aligns with Phe and Trp residues (Phe87; CYP101A1 numbering given) and His100 with Tyr residues (Tyr96). However there are similarities with Thr173 aligning with Thr residues (Thr185) and Val284 and Val385 with Val residues (Val295 and Val396; Table S2†).25,26 Additionally residues such as Thr85 and Ile384 (Thr101 and Ile395) align with other Thr and Ile residues in all the CYP101 family members except CYP101C1.16
In CYP101B1 there are also changes to the residues that comprise the potassium binding site and salt bridge network of CYP101A1. Glu84 and Tyr96 of the CYP1011A1 potassium binding site are replaced with Arg72 and His80, respectively in CYP101B1. In the salt bridge network most of the residues are conserved but there are modifications with Asp97 being replaced by Gly81 and Lys178 by Arg166. These differences when taken together with those in active site are likely to account for the lower affinity for camphor and the altered substrate profile and C–H bond oxidation selectivity of CYP101B1.5 It is of note that the number of different active site residues in CYP101B1 compared to CYP101A1 is greater than in both CYP101D enzymes but fewer when compared to CYP101C1 (Table S2† and Fig. 1 and 2).16
 | ||
| Fig. 2 A phylogenetic tree (phenogram) of the CYP101 family of P450 enzymes from N. aromaticivorans DSM12444, CYP101B1, CYP101C1, CYP101D1 and CYP101D2, CYP101A1 (P450cam) from Pseudomonas putida, and CYP101B2 from Novosphingobium tardaugens NBRC 16725. Included for comparison are P450cin (CYP176A1) the closest structurally characterised homologue to CYP101B1 outside of the CYP101 family. Also included are CYP109D1 and CYP264B1 from Sorangium cellulosum and CYP105D1 from Streptomyces griseus all of which can bind and catalyse the oxidation of norisoprenoids. | ||
The acid alcohol pair of the I-helix, which is involved in proton delivery and oxygen activation, and the glutamate and arginine pair (EXXR), which is proposed to be involved in heme incorporation, are conserved in CYP101B1.27 The normally conserved phenylalanine amino acid seven residues towards the N-terminus from the heme iron ligating cysteine, Cys346, is also present (Fig. 1).28 One potentially interesting point of difference in the CYP101B1 sequence is the presence of a proline residue, Pro347, one residue to the C-terminal side of the proximal cysteine. Proline occurs naturally at this position in chloroperoxidase and P450s such as CYP7A1, and CYP121A1.27,29,30 The introduction of proline residues at this position is known to have distinctive effects in other CYP enzymes, with the Leu358Pro mutant of CYP101A1 exhibiting the same spectral perturbations induced by putidaredoxin binding.31,32 The Ile401Pro mutant of CYP102A1 (P450Bm-3) displays enhanced oxidation rates with non-natural substrates and exhibits unusual electronic properties and structural changes.33,34
The substrate range of CYP101B1
Previously we have shown that β-ionone induces a large type I spin state shift (≥95%) on substrate binding (Kd, 0.23 ± 0.1 μM) to CYP101B1.5,9 The activity of CYP101B1 has been reconstituted using Arx, which is genomically associated with CYP101D2, in conjunction with ArR. Under standard assay conditions (0.5 μM CYP: 5 μM Arx) CYP101B1 oxidised β-ionone with high NADH consumption (1600 ± 100 nmol nmol P450−1 min−1, henceforth abbreviated to min−1) and product formation rates (1010 ± 60 min−1). This indicates that β-ionone may be, or is at least a close structural analogue of, the physiological substrate of CYP101B1. In this work similar spin state shifts and dissociation constants are observed with the closely related substrate α-ionone, ≥95% and 0.26 ± 0.04 μM (Fig. 3 and 4). The norisoprenoid β-damascone, which switches the position of the ketone and alkene moieties on the butenone side chain, displayed a lower spin state shift and weaker binding, 80% and 8.3 ± 0.9 μM (Fig. 3 and 4). Both substrates yielded fast NADH turnover frequencies with CYP101B1, α-ionone (1380 ± 140 min−1) and β-damascone (931 ± 13 min−1), though both are slower than β-ionone (Table 1).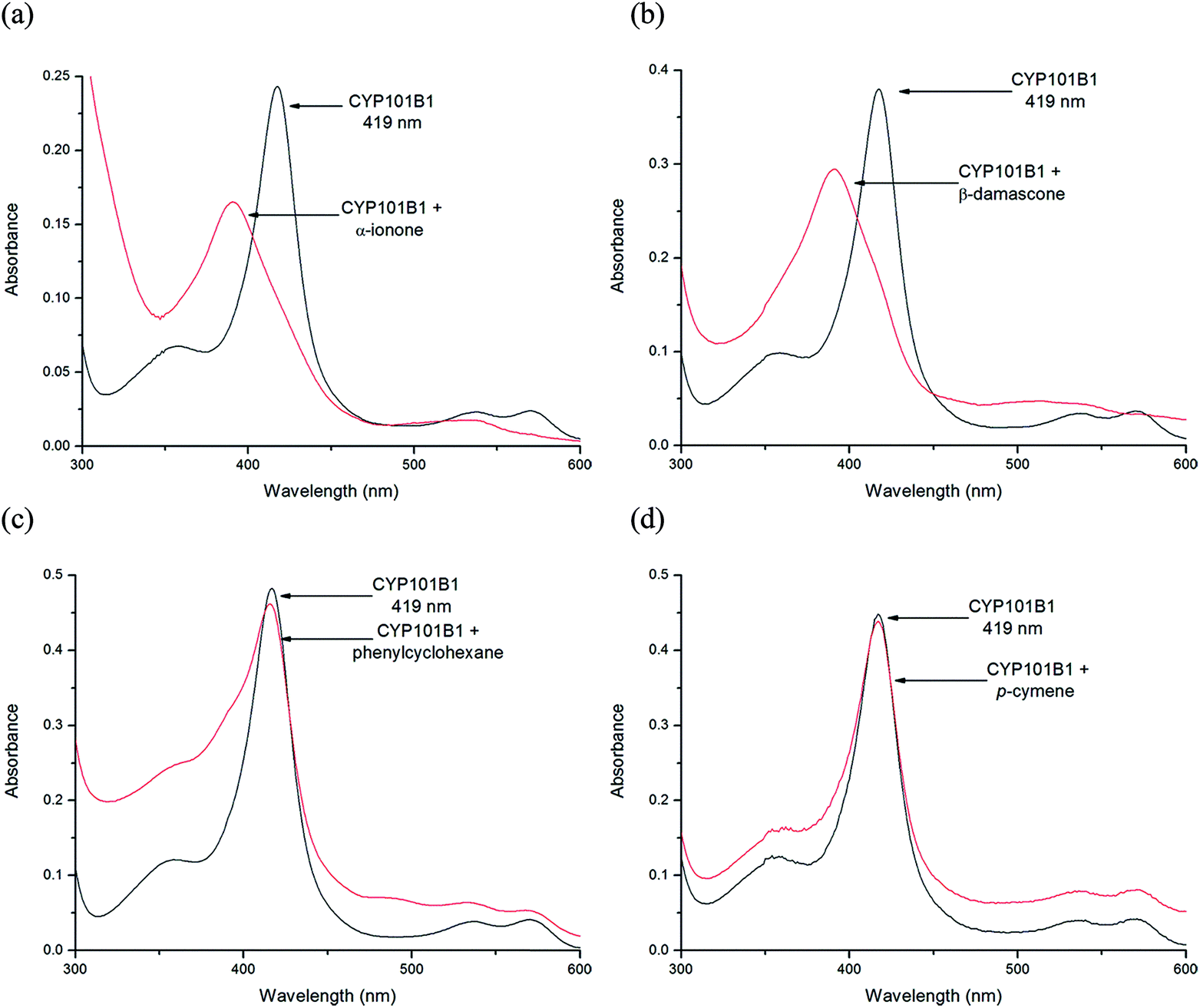 | ||
| Fig. 3 UV/Vis analysis of the CYP101B1 spin state shift with (a) α-ionone (b) β-damascone (c) phenylcyclohexane and (d) p-cymene. | ||
 | ||
| Fig. 4 Substrate dissociation constant analysis of (a) α-ionone, (b) β-damascone, (c) phenylcyclohexane and (d) p-cymene with CYP101B1. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Arx:CYP101B1 concentration ratio of 1:10:2 (0.5 μM CYP enzyme, 50 mM Tris, pH 7.4). Coupling is the percentage efficiency of NADH utilisation for the formation of products. Rates are reported as mean ± S.D. (n ≥ 3) and given in nmol nmol CYP−1 min−1. %HS is a measure of the shift in the spin state from low spin to high spin on substrate binding and Kd is the dissociation constant
:Arx:CYP101B1 concentration ratio of 1:10:2 (0.5 μM CYP enzyme, 50 mM Tris, pH 7.4). Coupling is the percentage efficiency of NADH utilisation for the formation of products. Rates are reported as mean ± S.D. (n ≥ 3) and given in nmol nmol CYP−1 min−1. %HS is a measure of the shift in the spin state from low spin to high spin on substrate binding and Kd is the dissociation constant
| CYP101B1/substrate | %HS heme | Kd (μM) | NADH consumption frequency (min−1) | Product formation rate (min−1) | Coupling (%) |
|---|---|---|---|---|---|
| β-Ionone | ≥95% | 0.23 ± 0.1 | 1600 ± 100 | 1010 ± 60 | 63 |
| α-Ionone | ≥95% | 0.26 ± 0.04 | 1380 ± 140 | 663 ± 56 | 48 |
| β-Damascone | 80% | 8.3 ± 0.9 | 931 ± 13 | 562 ± 12 | 60 |
| Phenylcyclohexane | 20% | 7.8 ± 0.9 | 293 ± 9.0 | 141 ± 17 | 48 |
| p-Cymene | 5% | 23.0 ± 3.0 | 197 ± 27 | 25.3 ± 6.0 | 13 |
The oxidation of β-damascone generated one major product (∼86%) and two minor products (Scheme 1 and Fig. 5). GC analysis revealed two major products from α-ionone oxidation in 63:37% ratio (Fig. 5). The product formation rate of α-ionone and β-damascone were similar, 663 ± 56 min−1 and 562 ± 12 min−1, with the coupling of NADH to product formation for β-damascone (60%) compensating for its lower NADH turnover frequency when compared to α-ionone (Table 1).
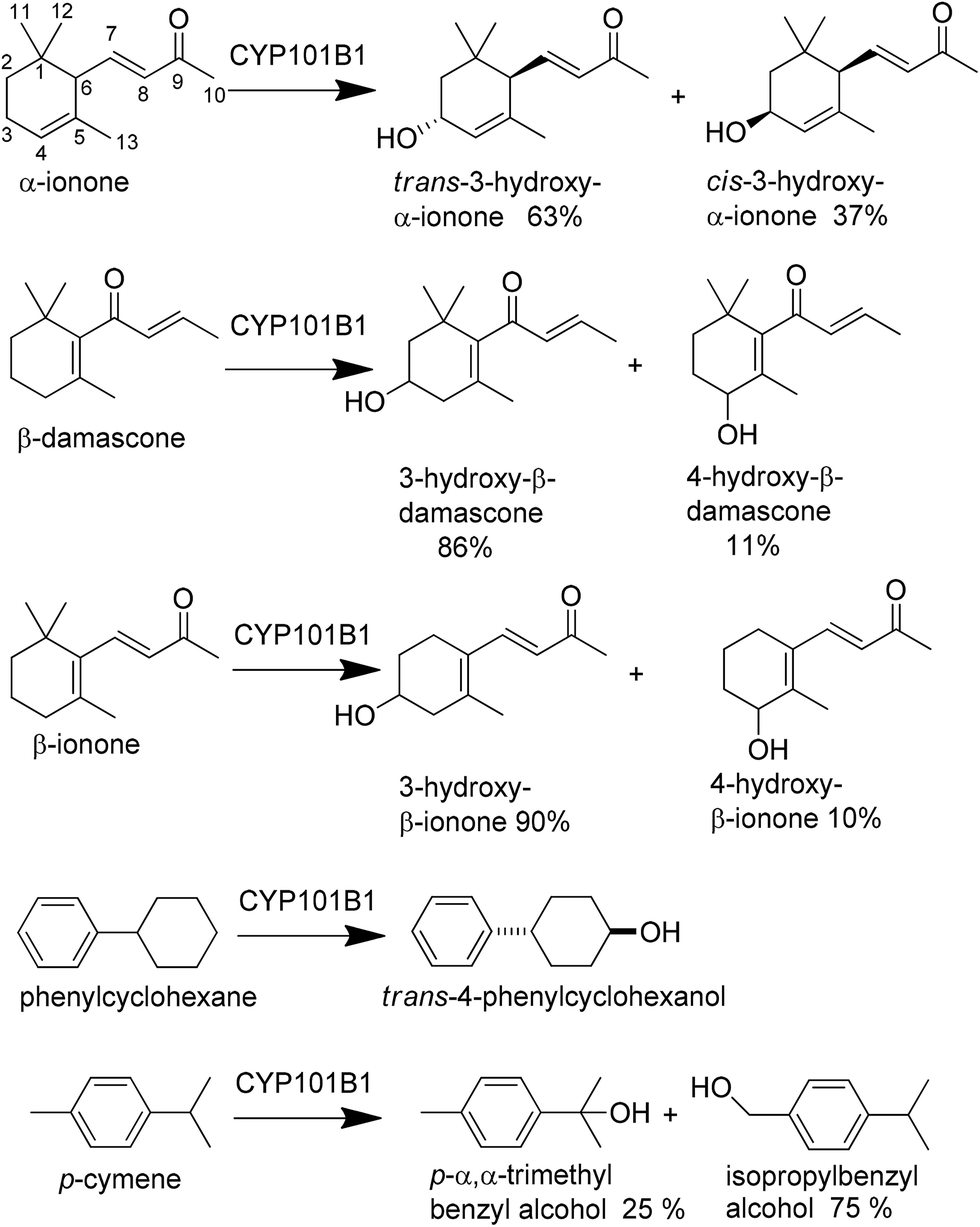 | ||
| Scheme 1 Substrate tested with CYP101B1 and the products isolated from the turnovers. | ||
 | ||
| Fig. 5 GC-MS (and HPLC) analysis of CYP101B1 turnovers (Substrate, internal standard (IS) and impurities (*) are labeled); (a) in vitro turnover of α-ionone, (b) in vivo turnover of α-ionone (A, trans-3-hydroxy-α-ionone, B, cis-3-hydroxy-α-ionone and C, 3-oxo-α-ionone). (c) in vitro turnover of β-ionone (A, 4-hydroxy-β-ionone and B 3-hydroxy-β-ionone). (d) HPLC analysis of the in vitro turnover of CYP101B1 and β-damascone; A; 3-hydroxy-β-damascone, B; 4-hydroxy-β-ionone and C; a third minor oxidation product (likely a ketone). | ||
Using a whole-cell oxidation system which produced CYP101B1, Arx and ArR in E. coli we were able to generate 100–200 mg of a mixture of the products formed from each norisoprenoid. The whole-cell biotransformation of α-ionone generates three products all in roughly equal amounts (30:37:33% ratio, Fig. 5). The first two of these correspond to the two products observed in vitro and have masses indicating they are hydroxylated monooxygenase products (Mass 208.2 AMU). The third product, which was not observed in the in vitro turnovers, has a mass indicating it arises from further oxidation of an alcohol product to a ketone (Mass 206.1 AMU). The whole-cell oxidation of β-damascone and β-ionone yielded predominantly one main product (Fig. 5) which corresponded to the products of the in vitro turnover systems. β-Damascone oxidation produced two minor products in low yields. Purification and isolation of the individual products was carried out using silica gel chromatography with only the fractions containing predominantly a single compound being taken forward for characterisation. We isolated between 2–10 mg pure product from each turnover of β-ionone, α-ionone and β-damascone after column chromatography, with the remaining product being found in mixed fractions. These pure samples were used to identify the products using NMR and mass spectrometry (Experimental section and ESI†). The products of α-ionone oxidation were identified as cis- and trans-3-hydroxy-α-ionone and 3-oxo-α-ionone (Scheme 1). The lack of significant quantities of the further oxidation product 3-oxo-α-ionone in the in vitro enzyme assays suggests it is only produced when the amount of α-ionone substrate is limited relative to the two hydroxylated products or that the increased solubility of the alcohols allow them to enter the cells more readily than the less soluble substrate. Furthermore the proportion of the cis and trans isomers were different in the whole-cell oxidation turnover suggesting that the trans isomer is more readily converted to 3-oxo-α-ionone. The regioselective oxidation of α-ionone at C3 and the absence of any evidence of the epoxidation of the double bond suggests that the substrate must be held in the active site with both the C3 pseudoequatorial and pseudoaxial C–H bonds held close to the heme iron.
β-Damascone oxidation by CYP101B1 yielded 86% of the majority product, 3-hydroxy-β-damascone, and 11% of the more abundant minor product, 4-hydroxy-β-damascone (Scheme 1). The other minor product that was produced (≤3% of an in vitro reaction) was not generated in a sufficiently pure enough form in high enough yields for NMR characterisation. GC-MS analysis indicated that it is likely to be a further oxidation product of one of the alcohols (Mass 206.2 AMU). These results are similar to those obtained previously with β-ionone. The major product from β-ionone was confirmed by NMR as 3-hydroxy-β-ionone and the minor product co-eluted with 4-hydroxy-β-ionone produced by CYP101C1 and CYP102A1.9,16,35 The ratio of 3-hydroxy- to 4-hydroxy-β-damascone is similar to that observed for the equivalent products of β-ionone suggesting these two substrates are bound in a similar orientation in the active site with the C3 carbon being closest to the heme iron followed by C4.
When using the CYP101B1 whole-cell oxidation system we previously observed the formation of a blue coloured pigment in cells after induction, which can be the result of indigo formation from indole oxidation.9 Here we show that the addition of indole to this system resulted in increased levels of blue pigment (Fig. S1†). This indicates that indole and related aromatic molecules may be viable substrates for oxidation by CYP101B1. Therefore we tested a small selection of aromatic substrates for their ability to bind to and be oxidised by CYP101B1 (indole, p-cymene and phenylcyclohexane). p-Cymene resulted in virtually no shift to the high spin form compared to indole which induced a small spin state shift (15%). Phenylcyclohexane induced a slightly larger shift of 20% but bound to CYP101B1 with comparable affinity to β-damascone (Table 1, Fig. 3 and 4).
Both phenylcyclohexane and p-cymene induced moderate activity with CYP101B1 as measured by the NADH turnover frequency, but both are lower when compared to the norisoprenoids. Phenylcyclohexane was more active, 293 ± 9.0 min−1, than p-cymene, 197 ± 6.0 min−1. Indole did not induce any enzyme activity above that of the leak rate. HPLC and GC analysis of phenylcyclohexane turnover indicated that a single product was formed (Fig. 6). This was isolated using the whole-cell oxidation system, purified (∼10 mg) and characterised using NMR as trans-4-phenylcyclohexanol. Coupling of the NADH consumption to product formation was calculated to be 48% resulting in a product formation rate of 141 ± 17 min−1. Two products were observed in small quantities in the turnover of p-cymene. These were identified via GC and HPLC co-elution experiments as p-α,α-trimethylbenzyl alcohol (25%) and isopropylbenzyl alcohol (75%). The coupling of NADH to product formation and the product formation rates are low (13% and 25.3 ± 6.0 min−1).
 | ||
| Fig. 6 GC-MS analysis of CYP101B1 turnovers; (a) in vitro turnover of phenylcyclohexane (A, trans-4-phenylcyclohexanol) and (b) in vitro turnover of p-cymene (A, p-α,α-trimethylbenzylalcohol and B, isopropylbenzyl alcohol). Substrate, internal standard (IS) and impurities (*) are labeled. | ||
Overall the data suggests certain aromatic substrates such as phenylcyclohexane are good substrates for CYP101B1 resulting in moderate activity and regioselective hydroxylations. However the norisoprenoid type substrates, ionone and damascone, show high affinities and activities for CYP101B1 and are efficiently oxidised with high regioselectivity and rates of product formation. α-Ionone is solely oxidised at the allylic C3 C–H compared to β-ionone and β-damascone where C3 oxidation dominates but with a contribution from allylic C4 hydroxylation.
Discussion
To date the CYP101B1 enzyme from N. aromaticivorans is one of only two members of this subfamily of P450 enzymes known with the other member only having 59% sequence identity. The low number of CYP101B subfamily members and the presence of both in Novosphingobium species hint at a potentially unique functional role. It is of note that CYP101B1 is located on a plasmid, whereas the electron transfer partners, ArR and Arx are located on the chromosomal genomic DNA with CYP101D1, CYP101D2 and CYP101C1.5,9 The active site residues of CYP101B1 vary when compared to those of the camphor oxidising CYP101A1, CYP101D1 and CYP101D2 and these differences will modify the substrate selectivity away from camphor towards alternative substrates such as norisoprenoids. The most significant change is the absence of an equivalent residue to tyrosine 96 of CYP101A1 which in CYP101B1 is replaced with a histidine residue.Potassium ions have been shown to increase the strength of camphor binding to CYP101A1. The behaviour of CYP101 family enzyme from N. aromaticivorans is different due to changes in the residues that comprise the camphor binding site between the CYP101 family members. The K+-binding site in CYP101A1 is octahedral, with the backbone carbonyl oxygens of Glu84, Gly93, Glu94 and Tyr96, and two water molecules, as ligands.36 These residues are conserved in CYP101D2, but not in CYP101D1, CY101C1 and CYP101B1. As well as the change at Tyr96, Glu84, is replaced with Arg residues in CYP101B1 and CYP101D1 and Gln in CYP101C1 which significantly reduces the effect of potassium on substrate binding.14 We have previously reported that, in common with these other CYP101 enzymes, CYP101B1 shows decreased activity at higher ionic strength predominantly due to an increasing KM.14 The proximal face of CYP101B1 is likely to consist of more positively charged than negatively charged residues, similar to CYP101C1, CYP101D1 and CYP101D2, allowing for strong interactions with the negatively charged surface potential of Arx in the vicinity of the [2Fe–2S] cluster.14
The side-chains of Lys178, Asp182, Thr185, Arg186, Asp251, and Asn255 are involved in the extensive salt bridge interactions in camphor-bound CYP101A1.37,38 In addition the Asp97 (B′ helix) and Lys197 (G helix) interaction has been proposed to play a role in camphor entry and product exit. Structural analysis of CYP101D1, CYP101D2 and CYP101C1 revealed that this salt bridge network was less extensive in these CYP101 family members. In CYP101B1 the changes of Lys178 to an arginine and the more dramatic alteration of Asp97 to a glycine would also be expected to alter this salt bridge network. The modifications in these regions along with those in the active site will alter the substrate range and C–H bond oxidation selectively of CYP101B1, relative to the other CYP101 subfamily members. Overall these changes in CYP101B1, when compared with CYP101A1, are fewer than those of CYP101C1 which agrees with the observation that CYP101B1 retains some binding affinity for camphor while binding of this substrate is virtually abolished in CYP101C1.5,9
CYP101B1 is able to bind norisoprenoids and camphor as well as selected substituted aromatics. It is tempting to speculate that the proline residue one position to the C-terminus of the proximal iron binding cysteinate residue contributes to the seemingly broader substrate range of this enzyme, though this remains to be confirmed. This would agree with data from the Ile401Pro variant of CYP102A1 but the CYP121A1 enzyme from Mycobacterium tuberculosis which has a proline in the same position has been reported to have a very specific substrate and reaction type, the C–C coupling of a cyclodipeptide.39
The physiological substrate of CYP101B1 is yet to be confirmed. However, the norisoprenoid class of compounds such as β- and α-ionone and β-damascone are efficiently oxidised, the binding affinity for these substrates is relatively strong and they induce large type I spin state shifts. Norisoprenoids can be derived from the breakdown of carotenoids and N. aromaticivorans contains at least one carotenoid dioxygenase, though this was identified as a 13,14-dioxygenase.40 The relative location of the ketone and alkene functionalities in the butenone side chain has more of an effect on the binding affinity than the location of the alkene double bond in the cyclohexyl ring with α- and β-ionone being better substrates than β-damascone. The location of the double bond in the cyclohexyl ring does have an effect on the selectivity of the hydroxylation. The allylic C3 C–H bonds of α-ionone being exclusively oxidised but for β-ionone and β-damascone allylic C4 hydroxylation is observed alongside C3 oxidation. The C–H bonds of the C3 in the norisoprenoid substrates, which are oxidised in the major products, are presumably held closer to the heme iron than any other C–H bond. The lower regioselectivity of β-ionone and β-damascone is presumably due to the more reactive hydrogens on the allylic C4 being in close enough proximity to the heme iron enabling them to compete with oxidation at C3. In α-ionone the C4 hydrogen is vinylic and the reactive alkene double bond must be too far away from the heme iron for epoxidation to occur.
Microbial oxidative biotransformations of α- and β-ionones have been reported using different bacteria including Streptomyces.21 P450 enzymes from S. griseolus and S. griseus were shown to oxidise α- and β-ionone at the allylic C3 and C4 positions, respectively.41 Variants of the self-sufficient cytochrome P450, CYP102A1, are able to selectively oxidise β-ionone at the allylic C4 position.35 Others have reported the stereoselective oxidation of the different enantiomers of α-ionone at the C3 position.42 The best reported product formation rates and coupling of these variants of the highly efficient and self-sufficient CYP102A1 enzyme with norisoprenoids are 280 min−1 and 35% for β-ionone, 152 min−1 and 39% for (R)-α-ionone and 98 and 44% for (S)-α-ionone, which are lower than those observed with the CYP101B1/ArR/Arx system.42
Other researchers have reported the binding and oxidation of ionones by the CYP264B1, CYP109B1 and CYP109D1 bacterial enzymes. The CYP109 enzymes were regioselective for oxidation at the allylic position while CYP264B1 oxidised both α- and β-ionones solely at C3.20,23,43 Non-physiological electron transfer partners were used for all three of these cytochrome P450 enzymes and the catalytic rates were slow compared to those reported for CYP102A1 and CYP101B1 (<5 min−1).
CYP101C1 from the same Novosphingobium bacterium is interesting in that it can rapidly oxidise β-ionone (PFR ∼2000 min−1) to a mixture of 4-hydroxy- (75%) and 3-hydroxy-β-ionone (25%) but the turnover with α-ionone is much slower (PFR ∼ 220 min−1). CYP101C1 oxidation of α-ionone is also regioselective for allylic C3 oxidation. It can also oxidise β-damascone to a mixture of 3- and 4-hydroxy products as well as further oxidation products. CYP101B1 is more active than all the other P450s tested so far for α-ionone oxidation. Whether this reaction is stereoselective with enantiomerically pure α-ionones remains to be determined. With β-ionone and β-damascone CYP101B1 is less active than CYP101C1 but alters the selectivity to favour oxidation predominantly at the C3 rather than the allylic C4 position. Of the other enzymes utilised to oxidise ionones, CYP102A1 mutants and the CYP109 enzymes family are less active but are more selective for allylic hydroxylation at C4 while CYP264B1 is very selective for oxidation at C3 but has very low activity.
The selective oxidation of the unrelated and more hydrophobic aromatic substrate phenylcyclohexane to trans-4-phenylcyclohexanol by CYP101B1 on the aliphatic ring at moderate activity is interesting. Tyrosine variants of CYP101A1 have been used to oxidise phenylcyclohexane at predominantly the 4-position though the activities were lower. Other mutations in the active site were also shown to alter the selectivity though none were completely regioselective.44–46 CYP108D1, also from N. aromaticivorans, has also been reported to tightly bind phenylcyclohexane and oxidise it to trans-4-phenylcyclohexanol, though the activity using non-physiological electron transfer partners is very low.47 The oxidation of p-cymene was significantly slower than phenylcyclohexane suggesting the smaller size of this substrates is detrimental to binding and productive and selective turnover. p-Cymene can bind with either methyl or isopropyl substituent closest to the heme iron. The combination of selective oxidations, high monooxygenase activities and the large total number of turnovers obtained using the whole-cell oxidation system make CYP101B1 ideally suited to the biocatalytic generation of hydroxylated products.
Experimental section
General
General reagents and organics were from Sigma-Aldrich or VWR, Australia. Buffer components, NADH, and isopropyl-β-D-thiogalactopyranoside (IPTG) were from Anachem (Astral Scientific, Australia) or Biovectra, (Scimar, Australia). UV/Vis spectroscopy was performed on Varian Cary 5000 or Agilent Cary 60 spectrophotometers. Gas chromatography (GC) analyses were carried out on a Shimadzu GC-17A instrument coupled to a QP5050A MS detector using a DB-5 MS fused silica column (30 m × 0.25 mm, 0.25 μm) and helium as the carrier gas. Analytical liquid chromatography was performed using an Agilent 1260 Infinity pump equipped with an Agilent Eclipse Plus C18 column (250 mm × 4.6 mm, 5 μm), an autoinjector and UV detector. A gradient, 20–95%, of acetonitrile (with trifluoroacetic acid, 0.1%) in water (TFA, 0.1%) was used. NMR spectra were recorded using a Varian Inova-600 spectrometer.Phylogenetic analysis
BLAST searches were performed using the databases at the National Center for Biotechnology Information. Sequence alignments were performed using ClustalW.48 The evolutionary history was inferred by using the Maximum Likelihood method based on the Jones Taylor Thornton (JTT) matrix-based model.49 The tree with the highest log likelihood is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 386 positions in the final dataset. Evolutionary analyses were conducted in MEGA6.50Protein production and purification
General DNA manipulations and microbiological experiments and the expression, purification and quantitation of the electron transfer proteins ArR and Arx from N. aromaticivorans were performed as described elsewhere.5,9 The CYP101B1 gene has been cloned into the pET26 vector and the encoded CYP101B1 protein was produced in E. coli BL21 (DE3).5,9,51 A single colony was inoculated into 20 mL Luria–Bertani broth (LB) containing kanamycin (25 mg L−1; LBkan) and grown at 37 °C for 6 hours. This culture was then inoculated into LBkan (1 mL per 500 mL) medium and grown at 37 °C for 6 h to an OD600 of ∼1. Protein production was induced by the addition of IPTG (0.1 mM, from a 0.5 M stock in water). The incubator temperature was lowered to 25 °C, and the culture was grown for a further 16 h. The red cell pellet, harvested from the culture by centrifugation (9000g), was resuspended in 200 mL of buffer T (50 mM Tris, pH 7.4). The cells were lysed by sonication (30 × 20 s pulses with 40 s gap in between each pulse) and the cell debris was removed by centrifugation 27000g for 30 min at 4 °C. The supernatant was loaded onto a DEAE Fastflow Sepharose column (200 mm × 50 mm; GE Healthcare) and the protein eluted using a linear salt gradient of KCl (80–350 mM) in buffer T developed over eight column bed-volumes at a flow rate of 8 mL min−1. The red protein-containing fractions were collected and concentrated by ultrafiltration. The protein was desalted using a gel filtration step on a G25 Sephadex column (600 mm × 10 mm) using Buffer T. The final purification step was anion-exchange chromatography using a Source-15Q column (120 mm × 26 mm; GE Healthcare) and eluting with a linear gradient of 50–250 mM KCl in buffer T developed over 20 column bed-volumes at a flow rate of 6 mL min−1. Purified fractions (A418/A280 ≥ 1.2) were combined and concentrated by ultrafiltration. An equal volume of glycerol was added, the solution filtered through a 0.22 μm sterile syringe filter and stored at −20 °C. CYP101B1 was quantitated using ε417 = 113 mM−1 cm−1.9
Substrate binding and kinetic analysis
For substrate binding the P450 enzymes were diluted to ∼2.0 μM using 50 mM Tris, pH 7.4. The high spin heme content was estimated, to approximately ±5%, by comparison with a set of spectra generated from the sum of the appropriate percentages of the spectra of the substrate-free form (>95% low spin, Soret maximum at 418 nm) and camphor-bound form (>95% high spin, Soret maximum at 392 nm) of wild-type CYP101A1.To determine the dissociation constant the CYP101B1 enzyme was prepared as above in a total volume 2.5 mL and used to baseline the spectrophotometer. Aliquots of substrate (0.5–2 μL) were added using a Hamilton syringe from a 1, 10 or 100 mM stock solution in ethanol. The solution was mixed and the peak-to-trough difference in absorbance recorded between 700 nm and 250 nm. Further aliquots of substrate were added until the peak-to-trough difference did not shift further. The apparent dissociation constants, Kd, were obtained by fitting the peak-to-trough difference against substrate concentration to a hyperbolic function (eqn (1)):
 | (1) |
Both ionone substrates exhibited tight binding, with Kd < 5[E], and in these instances the data were fitted to the tight binding quadratic equation (eqn (2)):
 | (2) |
NADH turnover assays were performed with mixtures (1.2 mL) containing 50 mM Tris, pH 7.4, 0.5 μM CYP101B1, 5 μM Arx, 1 μM ArR and 100 μg mL−1 bovine liver catalase. The mixtures were oxygenated and then equilibrated at 30 °C for 2 min. Substrates were added as 100 mM stock solutions in ethanol to a final concentration of 1 mM. NADH was added to ∼320 μM, final A340 = 2.00, and the absorbance at 340 nm was monitored. The rate of NADH turnover was calculated using ε340 = 6.22 mM−1 cm−1 (Table 1).
Product formation and isolation
Where authentic product standards were available they were utilised for identification purposes via HPLC and GC co-elution experiments. Extraction of the products from activity assays and product analysis by GC were performed as before.9 The p-cymene turnover products isopropylbenzyl alcohol and thymol eluted together on the GC-MS and HPLC. The alcohol products were derivatised using BSTFA/TMCS and analysed via GC-MS to identify the product as isopropyl benzyl alcohol rather than thymol. The product concentration in incubation mixtures was calculated by calibrating the total ion count response of the GC detector to products when available (or the substrate if the product was not available). Where more than one product was produced the detector response was assumed to be equal, e.g. 4-hydroxy-β-damascone and 3-hydroxy-β-damascone were assumed to have equal detector responses. The coupling efficiency was the percentage of NADH consumed that led to product formation.GC-MS analyses were carried out with the injector held at 250 °C and the interface at 280 °C. The oven temperature was varied as follows: for norisoprenoids and phenylcyclohexane, 120 °C for 3 minutes followed by a gradient of 10 °C min−1 for 10 min and held at 220 °C for a further 7 minutes; p-cymene 80 °C for 3 minutes followed by a gradient of 10 °C min−1 for 14 min and held at 220 °C for a further 3 minutes. The GC retention times were as follows: β-ionone, 7.5 min; 4-hydroxy-β-ionone, 9.9 min; 3-hydroxy-β-ionone, 10.3 min; α-ionone, 6.7 min; trans-3-hydroxy-α-ionone, 9.4 min; cis-3-hydroxy-α-ionone, 9.5 min; 3-oxo-α-ionone, 9.8 min; β-damascone, 6.5 min; 4-hydroxy-β-damascone, 9.1 min; 3-hydroxy-β-damascone, 9.3 min; phenylcyclohexane, 5.1 min; trans-4-hydroxyphenylcyclohexane, 7.7 min; p-cymene 4.4 min; isopropylbenzyl alcohol, 8.9 min (14.0 after derivatisation); p-α,α-trimethylbenzylalcohol, 7.2 min; thymol, 8.9 min (11.9 after derivatisation); carvacrol, 9.1 min.
To isolate and identify products where standards were not available a whole-cell oxidation system utilising the plasmids pETDuetArx/ArR and pRSFDuetArx/CYP101B1 was used to oxidise β-ionone, α-ionone, β-damascone and phenylcyclohexane, as described previously.9 The supernatant (200 mL) was extracted in ethylacetate (3 × 100 mL), washed with brine (100 mL) and dried with magnesium sulphate and the organic extracts were pooled and the solvent was removed by vacuum distillation and then under a stream of nitrogen. The products were purified using silica gel chromatography using a hexane–ethyl acetate stepwise gradient ranging from 80:20 to 50:50 hexane to ethyl acetate using 2.5% increases every 100 mL. The composition of the fractions was assessed by TLC and GC-MS and those containing single products (≥95%) were combined for characterisation. The solvent was removed under reduced pressure. The purified product was dissolved in CDCl3 and the organics characterised by NMR spectroscopy and GC-MS. NMR spectra were acquired on a Varian Inova-600 and spectrometer operating at 600 MHz for 1H and 150 MHz for 13C.
A combination of 1H, 13C, COSY, HSQC and HMBC experiments was used to determine the structures of the products. The cis/trans configuration of 3-hydroxy-α-ionone diastereomers was attributed from the assignments of Yamazaki52 and the selective nOe difference measurements in ROESY experiments. The diagnostic nOe was between the H3 and H6 protons in the cis diastereomer with the C11 methyl 3H signal.
NMR and GC MS data
Conclusion
CYP101B1 is a valuable addition to the field of monooxygenase biocatalysis. It is stable, produced in high yield and its electron transfer partners are known. It has product formation rates that approach, and in certain cases exceed, those of the self-sufficient CYP102A1. The coupling of reducing equivalents to product formation is also high. Using a whole-cell oxidation system we were able to generate significant quantities of hydroxylated metabolites. CYP101B1 was capable of oxidising a broad range of substrates including norisoprenoids, monoterpenes and hydrophobic aromatic compounds, which is distinct to the other members of the CYP101 family and therefore offers a specialised substrate binding profile. Taken together these factors, and the additional potential to engineer CYP101B1 for more efficient and selective turnovers, will enable the development of biocatalytic routes to as yet unobtainable fine chemicals.Acknowledgements
The work was supported by the School of Chemistry & Physics at the University of Adelaide (to S.G.B.). E.A.H. thanks the University of Adelaide for an M. Phil Scholarship.References
- Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Kluwer Academic/Plenum Press, New York, 3rd edn, 2005 Search PubMed.
- The Ubiquitous Roles of Cytochrome P450 Proteins, ed. A. Sigel, H. Sigel and R. Sigel, John Wiley & Sons, 1st edn, 2007 Search PubMed.
- R. Bernhardt and V. B. Urlacher, Appl. Microbiol. Biotechnol., 2014, 98, 6185–6203 CrossRef CAS PubMed.
- T. Furuya and K. Kino, Appl. Microbiol. Biotechnol., 2010, 86, 991–1002 CrossRef CAS PubMed.
- S. G. Bell and L. L. Wong, Biochem. Biophys. Res. Commun., 2007, 360, 666–672 CrossRef CAS PubMed.
- F. Hannemann, A. Bichet, K. M. Ewen and R. Bernhardt, Biochim. Biophys. Acta, 2007, 1770, 330–344 CrossRef CAS PubMed.
- J. A. Peterson and S. E. Graham-Lorence, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Plenum Press, New York, 2nd edn, 1995, pp. 151–182 Search PubMed.
- S. G. Bell, F. Xu, E. O. Johnson, I. M. Forward, M. Bartlam, Z. Rao and L. L. Wong, J. Biol. Inorg. Chem., 2010, 15, 315–328 CrossRef CAS PubMed.
- S. G. Bell, A. Dale, N. H. Rees and L. L. Wong, Appl. Microbiol. Biotechnol., 2010, 86, 163–175 CrossRef CAS PubMed.
- C. Hong, S. G. Bell, W. Yang, H. Wang, Y. Hao, X. Li, W. Zhou, M. Bartlam and L. L. Wong, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2009, 65, 364–367 CrossRef CAS PubMed.
- R. Zhou, C. Huang, A. Zhang, S. G. Bell, W. Zhou and L. L. Wong, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 964–967 CrossRef CAS PubMed.
- J. K. Fredrickson, D. L. Balkwill, G. R. Drake, M. F. Romine, D. B. Ringelberg and D. C. White, Appl. Environ. Microbiol., 1995, 61, 1917–1922 CAS.
- J. K. Fredrickson, F. J. Brockman, D. J. Workman, S. W. Li and T. O. Stevens, Appl. Environ. Microbiol., 1991, 57, 796–803 CAS.
- W. Yang, S. G. Bell, H. Wang, W. Zhou, N. Hoskins, A. Dale, M. Bartlam, L. L. Wong and Z. Rao, J. Biol. Chem., 2010, 285, 27372–27384 CrossRef CAS PubMed.
- S. G. Bell, W. Yang, A. Dale, W. Zhou and L. L. Wong, Appl. Microbiol. Biotechnol., 2013, 97, 3979–3990 CrossRef CAS PubMed.
- M. Ma, S. G. Bell, W. Yang, Y. Hao, N. H. Rees, M. Bartlam, W. Zhou, L. L. Wong and Z. Rao, ChemBioChem, 2011, 12, 88–99 CrossRef CAS PubMed.
- W. Yang, S. G. Bell, H. Wang, W. Zhou, M. Bartlam, L. L. Wong and Z. Rao, Biochem. J., 2011, 433, 85–93 CrossRef CAS PubMed.
- S. Vohra, M. Musgaard, S. G. Bell, L. L. Wong, W. Zhou and P. C. Biggin, Protein Sci., 2013, 22, 1218–1229 CrossRef CAS PubMed.
- E. M. Gillam, L. M. Notley, H. Cai, J. J. De Voss and F. P. Guengerich, Biochemistry, 2000, 39, 13817–13824 CrossRef CAS PubMed.
- Y. Khatri, M. Girhard, A. Romankiewicz, M. Ringle, F. Hannemann, V. B. Urlacher, M. C. Hutter and R. Bernhardt, Appl. Microbiol. Biotechnol., 2010, 88, 485–495 CrossRef CAS PubMed.
- S. Lutz-Wahl, P. Fischer, C. Schmidt-Dannert, W. Wohlleben, B. Hauer and R. D. Schmid, Appl. Environ. Microbiol., 1998, 64, 3878–3881 CAS.
- Y. T. Meharenna, K. E. Slessor, S. M. Cavaignac, T. L. Poulos and J. J. De Voss, J. Biol. Chem., 2008, 283, 10804–10812 CrossRef CAS PubMed.
- T. T. Ly, Y. Khatri, J. Zapp, M. C. Hutter and R. Bernhardt, Appl. Microbiol. Biotechnol., 2012, 95, 123–133 CrossRef CAS PubMed.
- X. Chen, A. Christopher, J. P. Jones, S. G. Bell, Q. Guo, F. Xu, Z. Rao and L. L. Wong, J. Biol. Chem., 2002, 277, 37519–37526 CrossRef CAS PubMed.
- F. Xu, S. G. Bell, J. Lednik, A. Insley, Z. Rao and L.-L. Wong, Angew. Chem., Int. Ed., 2005, 44, 4029–4032 CrossRef CAS PubMed.
- S. G. Bell, C. F. Harford-Cross and L.-L. Wong, Protein Eng., 2001, 14, 797–802 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- J. P. Clark, C. S. Miles, C. G. Mowat, M. D. Walkinshaw, G. A. Reid, S. N. Daff and S. K. Chapman, J. Inorg. Biochem., 2006, 100, 1075–1090 CrossRef CAS PubMed.
- W. Tempel, I. Grabovec, F. MacKenzie, Y. V. Dichenko, S. A. Usanov, A. A. Gilep, H. W. Park and N. Strushkevich, J. Lipid Res., 2014, 55, 1925–1932 CrossRef CAS PubMed.
- K. J. McLean, P. Carroll, D. G. Lewis, A. J. Dunford, H. E. Seward, R. Neeli, M. R. Cheesman, L. Marsollier, P. Douglas, W. E. Smith, I. Rosenkrands, S. T. Cole, D. Leys, T. Parish and A. W. Munro, J. Biol. Chem., 2008, 283, 33406–33416 CrossRef CAS PubMed.
- S. Nagano, T. Tosha, K. Ishimori, I. Morishima and T. L. Poulos, J. Biol. Chem., 2004, 279, 42844–42849 CrossRef CAS PubMed.
- T. Tosha, S. Yoshioka, K. Ishimori and I. Morishima, J. Biol. Chem., 2004, 279, 42836–42843 CrossRef CAS PubMed.
- C. J. Whitehouse, S. G. Bell, W. Yang, J. A. Yorke, C. F. Blanford, A. J. Strong, E. J. Morse, M. Bartlam, Z. Rao and L. L. Wong, ChemBioChem, 2009, 10, 1654–1656 CrossRef CAS PubMed.
- C. J. Whitehouse, W. Yang, J. A. Yorke, B. C. Rowlatt, A. J. Strong, C. F. Blanford, S. G. Bell, M. Bartlam, L. L. Wong and Z. Rao, ChemBioChem, 2010, 11, 2549–2556 CrossRef CAS PubMed.
- V. B. Urlacher, A. Makhsumkhanov and R. D. Schmid, Appl. Microbiol. Biotechnol., 2006, 70, 53–59 CrossRef CAS PubMed.
- A. C. Westlake, C. F. Harford-Cross, J. Donovan and L. L. Wong, Eur. J. Biochem., 1999, 265, 929–935 CrossRef CAS.
- E. Deprez, N. C. Gerber, C. Di Primo, P. Douzou, S. G. Sligar and G. Hui Bon Hoa, Biochemistry, 1994, 33, 14464–14468 CrossRef CAS.
- C. Di Primo, E. Deprez, S. G. Sligar and G. Hui Bon Hoa, Biochemistry, 1997, 36, 112–118 CrossRef CAS PubMed.
- P. Belin, M. H. Le Du, A. Fielding, O. Lequin, M. Jacquet, J. B. Charbonnier, A. Lecoq, R. Thai, M. Courcon, C. Masson, C. Dugave, R. Genet, J. L. Pernodet and M. Gondry, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7426–7431 CrossRef PubMed.
- Y. S. Kim, E. S. Seo and D. K. Oh, Biotechnol. Lett., 2012, 34, 1851–1856 CrossRef PubMed.
- A. Celik, S. L. Flitsch and N. J. Turner, Org. Biomol. Chem., 2005, 3, 2930–2934 CAS.
- H. Venkataraman, S. B. A. d. Beer, D. P. Geerke, N. P. E. Vermeulen and J. N. M. Commandeur, Adv. Synth. Catal., 2012, 354, 2172–2184 CrossRef CAS.
- M. Girhard, T. Klaus, Y. Khatri, R. Bernhardt and V. B. Urlacher, Appl. Microbiol. Biotechnol., 2010, 87, 595–607 CrossRef CAS PubMed.
- P. A. England, D. A. Rouch, A. C. G. Westlake, S. G. Bell, D. P. Nickerson, M. Webberley, S. L. Flitsch and L. L. Wong, Chem. Commun., 1996, 357–358 RSC.
- N. E. Jones, P. A. England, D. A. Rouch and L. L. Wong, Chem. Commun., 1996, 2413–2414 RSC.
- S. G. Bell, D. A. Rouch and L. L. Wong, J. Mol. Catal. B: Enzym., 1997, 3, 293–302 CrossRef CAS.
- S. G. Bell, W. Yang, J. A. Yorke, W. Zhou, H. Wang, J. Harmer, R. Copley, A. Zhang, R. Zhou, M. Bartlam, Z. Rao and L. L. Wong, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 277–291 CAS.
- R. Chenna, H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins and J. D. Thompson, Nucleic Acids Res., 2003, 31, 3497–3500 CrossRef CAS PubMed.
- D. T. Jones, W. R. Taylor and J. M. Thornton, Comput. Appl. Biosci., 1992, 8, 275–282 CrossRef CAS PubMed.
- K. Tamura, G. Stecher, D. Peterson, A. Filipski and S. Kumar, Mol. Biol. Evol., 2013, 30, 2725–2729 CrossRef CAS PubMed.
- E. M. Gillam, A. M. Aguinaldo, L. M. Notley, D. Kim, R. G. Mundkowski, A. A. Volkov, F. H. Arnold, P. Soucek, J. J. DeVoss and F. P. Guengerich, Biochem. Biophys. Res. Commun., 1999, 265, 469–472 CrossRef CAS PubMed.
- Y. Yamazaki, Y. Hayashi, M. Arita, T. Hieda and Y. Mikami, Appl. Environ. Microbiol., 1988, 54, 2354–2360 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14010a |
| This journal is © The Royal Society of Chemistry 2015 |