
Phthalocyanine–titanate nanotubes: a promising nanocarrier detectable by optical imaging in the so-called imaging window†
Abstract
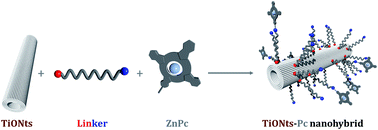
TiONts–phthalocyanine nanohybrids combining an efficient optical probe and a promising nanovector have been developed in a step-by-step approach and were thoroughly characterized. Each 150 nm long TiONts–Pc bear ca. 450 Pc. Three nanohybrids were prepared including three different linkers in quest for the best stability.
 Please wait while we load your content...
Please wait while we load your content...

