
Pseudopeptide Foldamers designed for photoinduced intramolecular electron transfer†
Abstract
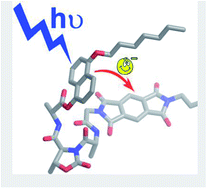
We have designed and prepared three pseudopeptide foldamers, called dyads 1, 2 and 3, equipped with a donor and an acceptor unit to promote intramolecular electron transfer after light excitation. All the three dyads contain the same donor and acceptor, which are a derivative of 1,5-dihydroxynaphthalene and a derivative of pyromellitic diimide, respectively. The donor and acceptor units are separated by hybrid foldamers of different length in order to vary both their distance and relative orientation. Specifically, one, two or three L-Ala-D-Oxd (Ala = alanine, Oxd = 4-carboxy-5-methyl-oxazolidin-2-one) units are contained in dyads 1, 2, and 3, respectively. Dyad 1 folds in a bent conformation in which the donor and acceptor units lie one close to the other, while dyads 2 and 3 preferentially assume an extended conformation. In all the three dyads both the donor and acceptor emissions are efficiently quenched via intramolecular electron transfer, as suggested by photophysical and electrochemical investigations. Because of its bent conformation dyad 1 exhibits a charge-transfer (CT) band at 410 nm in CH2Cl2 solution and a photoinduced electron transfer that occurs more efficiently than in dyads 2 and 3. Upon dissolving dyad 1 in DMSO, a competitive solvent for hydrogen bonds that establish in the pseudopeptide linker, the CT band disappears and the efficiency of electron transfer slightly decreases, in agreement with an unfolded conformation in which donor and acceptor units are no longer in close contact.
 Please wait while we load your content...
Please wait while we load your content...

