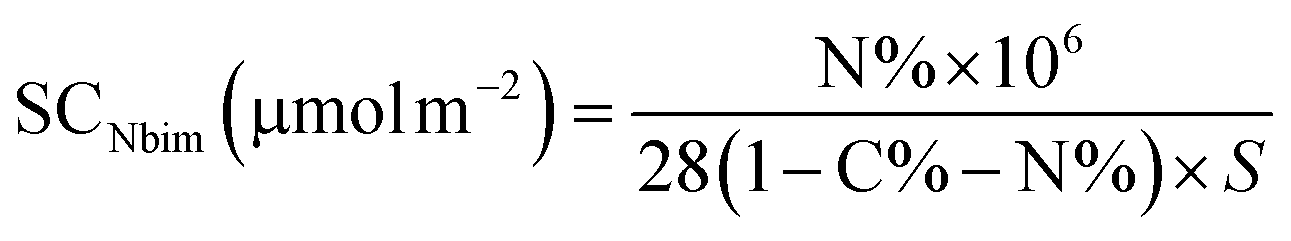DOI:
10.1039/C4RA13861A
(Paper)
RSC Adv., 2015,
5, 15500-15506
Nitro-substituted 3,3′-bis(indolyl)methane-modified silica gel as a sorbent for solid-phase extraction of flavonoids†
Received
5th November 2014
, Accepted 20th January 2015
First published on 20th January 2015
Abstract
A novel solid-phase extraction (SPE) sorbent was synthesized by chemical immobilization of nitro-substituted 3,3′-bis(indolyl)methane onto pure silica. The nitro-substituted 3,3′-bis(indolyl)methane-modified silica was evaluated by elemental analysis (EA) and Fourier transform infrared spectroscopy (FT-IR). Coupled to high performance liquid chromatography (HPLC), the extraction performance of the sorbent was evaluated by using five flavonoids as model analytes. The results showed that the new sorbent could offer multiple intermolecular interactions, such as π–π, hydrophobic, and hydrogen bonding interactions. Several factors including the extraction material, sample solution pH, sample loading rate, eluent type, volume of eluent, elution rate, and volume of sample loading were optimized. Under the optimal conditions, the proposed method was applied for the analysis of five flavonoids in grape juice. Satisfactory linear ranges for flavonoids were obtained in the range of 10–200 ng mL−1 for myricetin, 5–200 ng mL−1 for quercetin and apigenin, and 1–200 ng mL−1 for luteolin and kaempferol, with correlation coefficients (R) ranging from 0.9908 to 0.9996. Limits of detection (LODs) were in the range of 0.5–10 ng mL−1. The recovery values of spiked grape juice ranged from 91.5% to 120.7% with relative standard deviations (RSDs) less than 9.12% (n = 5). As a novel solid-phase extraction sorbent, the nitro-substituted 3,3′-modified silica exhibited a higher extraction efficiency towards the tested compounds than diol-modified silica.
1. Introduction
Polyphenolic compounds have recently gained considerable attention because of their antioxidant activity, anticancer and anti-inflammatory properties and their use against coronary heart disease (CHD).1–4 Polyphenolic compounds can be categorized into two large groups: flavonoids and non-flavonoids.5 To some extent, the protective effects mentioned above have been attributed to the antioxidant properties of flavonoids.6 The flavonoids can be further divided into six subclasses including flavonols, flavones, flavanones, flavanols, catechins, and anthocyanidins.7 Flavonols such as quercetin, myricetin, kaempferol and the corresponding flavones, apigenin and luteolin5 are important antioxidants, largely contributing to the prevention of oxidation of low-density lipoproteins and inhibiting lipid peroxidation.8,9 Considering the importance of the biological activity of flavonoids, developing an accurate and rapid method for their analysis is necessary.5 The occurrence of flavonoids in real samples is usually at a low concentration. Meanwhile, the sample matrices are generally complex, so it is imperative to select a sample pretreatment method before chromatographic analysis.10
Due to the advantages of a high enrichment factor, high recovery, rapid phase separation, simplicity of operation, low consumption of organic solvents, and ease of automation, solid-phase extraction (SPE) is now widely used for the isolation and concentration of various selected analytes from complex matrices.11–17 At present, an octadecyl (C18)-bonded phase has been widely used as an SPE sorbent for flavonoid sample preparation.18–20 However, the interaction between the C18-bonded phase and flavonoids was too simple, maybe only hydrophobic interactions, which resulted in certain compounds of a polarity similar to flavonoids interfering with the target compounds.21 Thus, the design and development of a novel sorbent according to the structure of flavonoids is required.
In this work, considering the polyphenolic structure of flavonoids, a new silica-based sorbent modified by a simple nitro-substituted 3,3′-bis(indolyl)methane (Nbim) compound bearing an amino group, indole ring and benzene ring, was designed and synthesized. The sorbent was used for solid-phase extraction of flavonoids mainly based on π–π, hydrophobic and hydrogen-bond interactions between the sorbent and flavonoids. Moreover, the introduction of electron-withdrawing nitro groups into the 3,3′-bis(indolyl)methane skeleton not only enhances the proton acidity of the H-bond donor site of the indole NH but also enhances the conjugation effect of the indole ring, therefore, the increased hydrogen-bonding and π–π interactions towards flavonoids could be expected. A control experiment using diol-modified silica as the sorbent was also carried out. Five flavonoids were selected as model analytes to evaluate the extraction performances of the sorbents by using HPLC analysis. The results showed that the introduction of Nbim into silica gel promoted the extraction efficiency by multiple intermolecular interactions between sorbent and flavonoids. Under the optimal conditions, the proposed method was effectively applied for the analysis of five flavonoids in grape juice.
2. Experimental
2.1 Chemicals and materials
5-Nitroindole was purchased from J&K Scientific Ltd. (Beijing, China). γ-(2,3-Epoxypropoxy)propyltrimethoxysilicane was purchased from Aladdin (Shanghai, China). Benzaldehyde and potassium hydrogen sulfate (KHSO4) were purchased from the Tianjin Chemical Reagent no. 2 Plant (Tianjin, China). NH3·H2O (28 wt%) and acetic acid (CH3COOH) were purchased from the Baiyin Liangyou Chemical Reagent Factory (Baiyin, China). Commercial silica (230–400 mesh) was purchased from Alfa Aesar (China). All solvents, except for methanol, were of analytical grade and were purchased from the Tianjin Chemical Reagent no. 2 Plant (Tianjin, China). Methanol (MeOH) was of chromatographic grade and was purchased from Qingdao Chemical Reagent Factory (Qingdao, China).
A set of analytes selected to evaluate the sorbent, i.e., myricetin (MYR), quercetin (QUR), luteolin (LUT), kaempferol (KAE), and apigenin (API) (as shown in Fig. 1), were all obtained from the Chengdu Must Bio-technology Co., Ltd. (Chengdu, China).
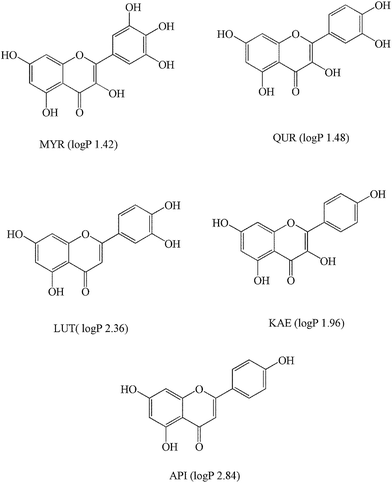 |
| | Fig. 1 The structures of the selected flavonoids. | |
2.2 Instrumentation and chromatographic conditions
The SPE procedure was performed on an 8-port model SPE Vacuum Manifold (Shanghai Hogon Scientific Instrument Co. Ltd.). SPE cartridges (3 mL) were purchased from Saifen-UCT Co. (Suzhou, China). Analysis of the model compounds was performed with an Agilent 1200 Series HPLC-DAD system equipped with a 20 mL sample loop. An AT. Lichrom C18 column (Hypersil ODS2, 250 mm × 4.6 mm I.D., 5 μm) was purchased from AT (Lanzhou, China). The mobile phase for the flavonoid compounds was composed of different proportions of (A) 0.25% phosphoric acid in water (acidified water) and (B) MeOH. The mobile phase was 40% A and 60% B. The flow-rate was 1.0 mL min−1 and the temperature of the column oven was set at 30 °C. The wavelength used to detect the flavonoid compounds was set at 360 nm.
Diffuse reflectance infrared Fourier transformation (DRIFT) spectra of the samples were obtained using a Thermo Nicolet 5700 FTIR spectrophotometer (Madison, WI, USA) in the range of 4000–400 cm−1. Elemental analysis was performed on an Elementar Vario EL (Hanau, Germany). The BET surface area, pore volume and average pore width of pure SiO2 and Nbim modified SiO2 were obtained on the nitrogen adsorption and desorption isotherms at 76.015 K using an ASAP2010 surface analysis instrument (Micromeritics, Norcross, GA 30093 USA).
2.3 Synthesis of Nbim
Nbim was synthesized according to a literature method.22 12.5 mM KHSO4 was added to a mixture of 25 mM 5-nitroindole and 12.5 mM benzaldehyde in dry MeOH under ultrasound. The mixture was refluxed for 12 h under nitrogen and cooled to room temperature afterwards. The precipitate was filtered, washed with MeOH several times and dried under vacuum at 60 °C, giving a yellow solid. The solid crude product was purified by recrystallization with acetone/H2O.
2.4 Preparation of the Nbim-modified silica sorbent and diol-modified silica sorbent
7.5 mM Nbim and 7.5 mM γ-(2,3-epoxypropoxy)propyltrimethoxysilicane were placed in a 100 mL reaction flask containing 25 mL dry methylethylketone. The mixture was refluxed for 24 h. Then, 40 mL toluene and 2.5 g silica were added into the reaction mixture in turn under ultrasound. The reaction mixture was stirred under nitrogen at 100 °C for 24 h. The modified silica was filtered and washed with toluene, ultrapure water and acetone in turn. The modified silica was characterized after being dried under vacuum. The preparation process of the Nbim-modified silica sorbent is described in Fig. 2a.
 |
| | Fig. 2 (a) Synthesis process of Nbim-modified silica. (b) Synthesis process of diol-modified silica. | |
The synthesis process of the diol-modified silica sorbent is schematically described in Fig. 2b. 7.5 mM γ-(2,3-epoxypropoxy)propyltrimethoxysilicane and 2.5 g silica were dispersed in 40 mL of toluene under ultrasound. The reaction mixture was stirred under nitrogen at 100 °C for 24 h. The obtained modified silica was washed with toluene, ultrapure water and ethanol. The diol-modified silica was characterized after being dried under vacuum.
The compound synthesized in the first step of Fig. 2a was confirmed by 1H NMR and 13C NMR (see ESI, Fig. S-1 and S-2†). 1H NMR (400 MHz, DMSO-d6, ppm): δ 11.74 (s, 1H), 8.38 (d, 2H), 8.03 (dd, 2H), 7.60 (d, 2H), 7.46 (d, 1H), 7.38 (t, 2H), 7.28 (t, 2H), 7.20 (s, 2H), 6.27 (s, 1H), 3.71 (m, 2H), 3.41 (s, 9H), 3.26 (m, 2H), 3.14 (m, 2H), 2.77 (t, 2H), 1.62 (s, 1H), 1.28 (s, 1H), 0.65 (t, 2H). 13C NMR (100 MHz, DMSO-d6): δ 144.2, 140.7, 140.2, 128.9, 128.7, 128.0, 126.8, 126.2, 120.9, 117.0, 116.7, 122.6, 72.9, 71.6, 50.8, 43.9, 22.9, 5.26.
2.5 Solid-phase extraction procedure
SPE experiments were carried out in the following way: 40 mg of Nbim-modified sorbent was packed into a 3 mL empty polypropylene cartridge equipped with two polyethylene frits, and then flushed with 5 mL of methanol and 5 mL water. 30 mL of sample solution (pH = 4) was passed through the cartridge at a flow rate of 1.5 mL min−1 as controlled by the adjustment of the vacuum during the whole SPE procedure. The retained analytes were eluted by 1 mL 1% acetic acid–methanol (v/v) at a flow rate of 0.5 mL min−1. The desorption solution was stored for HPLC analysis.
2.6 Preparation of standard solutions and sample collection
A series of stock standard solutions of each flavonoid for SPE were prepared in pure methanol at a concentration of 0.2 mg mL−1, and stored at 4 °C in a freezer. A multicomponent working solution was prepared daily by mixing and diluting the stock solutions with ultrapure water.
The grape juice drink was bought from a local supermarket. The grape juice samples were degassed by ultrasound and filtered through a vacuum filter (0.45 μm filter membrane) and then were stored at 4 °C before use.
3. Results and discussion
3.1 Characterization of the Nbim-modified silica sorbent and diol-modified silica sorbent
3.1.1 Elemental analysis. The elemental content was C 10.47%, H 2.08% and N 0.34% for the Nbim-modified silica sorbent, and C 9.79%, H 1.89% for the diol-modified silica sorbent. The surface coverage (SC), 0.28 μmol m−2 for Nbim and 6.16 μmol m−2 for diol, was calculated according to the following formulas:23
where C%, H%, and N% represent the percentage of carbon, hydrogen and nitrogen, respectively, and S is the specific surface area of the silica support (500 m2 g−1).
3.1.2 Infrared analysis. The successful modification of the silica was also confirmed by FT-IR analysis (Fig. 3). In the spectra of the Nbim-modified silica sorbent and the diol-modified silica sorbent, the broad band around 3448 cm−1, attributed to the stretching vibrations of O–H bonds of the geminal and vicinal silanols, decreased greatly, which was due to the decrease of silanols after bonding. Two new bands at 2930 and 2890 cm−1 were observed, which arise from the aliphatic C–H stretching vibration. In addition, in the spectrum of Nbim-modified silica sorbent, the new bands at 1520 and 1469 cm−1 were attributed to aromatic NO2 or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration modes.
C stretching vibration modes.
 |
| | Fig. 3 FT-IR of Nbim-modified silica (a), diol-modified silica (b) and pure silica (c). | |
3.1.3 Adsorption experiments. The BET surface area, total pore volume and average pore diameter of Nbim modified silica decreased (see ESI, Table S-1†) compared with pure silica, indicating that Nbim successfully modified the pure silica.
3.2 Optimization of the solid-phase extraction procedure
3.2.1 Extraction materials. It is important to select a SPE sorbent that can bind the compounds of interest.24 For all of the selected flavonoids, the recoveries of the Nbim-modified silica sorbent were higher than that of the diol-modified silica sorbent (Fig. 4). Considering the difference in the main structure between the two sorbents, the higher recovery of the Nbim-modified silica sorbent should be attributed to the introduction of Nbim, which could effectively enhance the π–π interaction and hydrogen bonding between the Nbim-modified silica sorbent and flavonoids. The π–π stack, hydrophobic, and hydrogen bonding interactions between the Nbim-modified silica sorbent and flavonoids played a predominant role in flavonoid extraction. Therefore, the Nbim-modified silica was chosen as the sorbent for extracting flavonoids in further research.
 |
| | Fig. 4 Effect of extraction material on the recovery of three parallel experiments. | |
3.2.2 Optimization of sample solution pH. The effect of sample solution pH (from 3.0 to 8.0) on the extraction performance of the Nbim-modified silica sorbent was investigated and the results are given in Fig. 5. Under different pH conditions, the order of the recoveries of the five selected flavonoids was mainly API > KAE > LUT > QUR > MYR, which related to the hydrophobic and hydrogen bonding interactions between the selected flavonoids and the Nbim-modified silica sorbent. Especially for MYR containing 6 phenolic hydroxyl groups (see Fig. 1), whose log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (octanol–water partition coefficient) was smaller than others, so the hydrophobic interaction between MYR and the Nbim-modified sorbent was weaker than others. Moreover, it was easier for MYR to form intramolecular hydrogen bonds, which weakened the intermolecular hydrogen bonds between MYR and the sorbent.
P (octanol–water partition coefficient) was smaller than others, so the hydrophobic interaction between MYR and the Nbim-modified sorbent was weaker than others. Moreover, it was easier for MYR to form intramolecular hydrogen bonds, which weakened the intermolecular hydrogen bonds between MYR and the sorbent.
 |
| | Fig. 5 Effect of sample solution pH on the recovery of three parallel experiments. | |
Flavonoids containing phenolic hydroxyl groups were acidic analytes, and their acidities were positively correlated to the number and position of the hydroxyl groups. The sample solution pH will not only strongly influence the state of the flavonoid analytes in solution as ionic or neutral molecules, but also affect the surface charge of the sorbent.25 As shown in Fig. 5, the recoveries of the selected flavonoids, especially MYR and QUR, increased with the increase of pH from 3 to 4 and then decreased as the pH increased from 4 to 8. When the sample solution pH increased, to some extent, the acidic flavonoids should suffer from ionization and exist in the form of anions, in addition, the Nbim-modified silica sorbent was also covered with negative charges, which could obviously weaken the adsorption capability for flavonoids due to the charge repulsive interaction between the sorbent and the flavonoid analytes. Thus, acidic conditions were needed to keep all the selected flavonoids stable in solution,26 and the optimum sample solution pH was found to be pH 4, at which satisfying recoveries were obtained for all the selected analytes.
3.2.3 Optimization of the sample loading rate. Generally, sample extraction time can be reduced by employing a high sample loading rate while possible extraction efficiency loss happened due to an incomplete adsorption of the target compounds by the sorbents.27 A sample loading rate ranging from 1.0 to 2.5 mL min−1 was evaluated. As shown in Fig. 6, for all of the selected flavonoids, the extraction recoveries increased with the sample loading rate increasing from 1 mL to 1.5 mL min−1, and then decreased from 1.5 mL min−1 to 2.5 mL min−1. When the sample loading rate was <1.5 mL min−1, some flavonoids adsorbed to the sorbent may have been eluted by the sample solution due to the hydrophilicity of flavonoids. When the sample rate was >1.5 mL min−1, an incomplete adsorption of flavonoids by the Nbim-modified silica sorbent happened. In order to achieve the maximum adsorption, 1.5 mL min−1 was selected as the sample loading rate.
 |
| | Fig. 6 Effect of the sample loading rate on the recovery of three parallel experiments. | |
3.2.4 Optimization of eluent type and volume. Eluent type and volume are vital for extraction efficiency. In this work, different eluents, including 1% acetic acid–methanol (v/v), 1% acetic acid–acetonitrile (v/v), 1% acetic acid–acetone (v/v), were tested as eluents for the investigation of eluting efficiencies of the selected flavonoids. However, when 1% acetic acid–acetonitrile (v/v) and 1% acetic acid–acetone (v/v) were selected as eluents, the solvent effects made the chromatographic peak of all the flavonoids undesirable and the tested flavonoids cannot be separated well, which made the quantitative analysis of the flavonoids difficult. Fig. 7 shows the recovery obtained from different volumes of 1% acetic acid–methanol (v/v), the recoveries of the flavonoid compounds increased with an increase in the volume of eluent from 0.5 mL to 1 mL. A further increase in volume of eluent resulted in little change in the recovery. So we chose a volume of 1 mL as the optimal eluent volume.
 |
| | Fig. 7 Effect of the volume of eluent on the recovery of three parallel experiments. | |
3.2.5 Optimization of the elution rate. The elution rate is another important factor which should be evaluated, because a high elution rate would result in incomplete desorption. As can be seen from Fig. 8, the recovery of most of the tested flavonoids decreased when the elution rate was higher than 0.5 mL min−1. Therefore, 0.5 mL min−1 was chosen as the elution rate.
 |
| | Fig. 8 Effect of the elution rate on the recovery of three parallel experiments. | |
3.2.6 Optimization of the volume of sample loading. Selection of an optimum volume of sample loading is essential for the sample pretreatment step and it is especially necessary for finding the maximum value of sample loading when we analyze the real sample. As shown in Fig. 9, the recovery of all the selected flavonoid compounds decreased greatly when the sample loading rate was larger than 30 mL. As a result, a 30 mL sample loading volume for flavonoid compounds was adopted.
 |
| | Fig. 9 Effect of the volume of sample loading on the recovery of three parallel experiments. | |
3.3 Analytical performance
Under the optimal conditions (see ESI, Table S-2†), the proposed SPE-HPLC-DAD method was investigated by testing the analytical parameters of flavonoid compounds in water samples. As shown in Table 1, good linearity for flavonoid compounds was obtained, which was in the range of 10–200 ng mL−1 for MYR, 5–200 ng mL−1 for QUR and API, and 1–200 ng mL−1 for LUT and KAE, with correlation coefficients (R) ranging from 0.9908 to 0.9996. The limits of detection (LODs, S/N = 3), investigated by extraction of ultrapure water samples spiked at different levels to meet such signal levels,28 were 5 ng mL−1 for MYR, 1 ng mL−1 for QUR and API, and 0.5 ng mL−1 for LUT and KAE. The limits of quantification (LOQs), the minimum concentration of the linear range, were 10 ng mL−1 for MYR, 5 ng mL−1 for QUR and API, and 1 ng mL−1 for LUT and KAE. The relative standard deviation (n = 5) for the 200 ng mL−1 standard solution of all the selected flavonoids was calculated to be less than 7.1%.
Table 1 Linear regression, LOD and LOQ of the developed method
| Flavonoids |
Linear range (ng mL−1) |
R2 |
LOD (ng mL−1) |
LOQ (ng mL−1) |
RSD (n = 5, %) |
| MYR |
10–200 |
0.9908 |
5 |
10 |
7.1 |
| QUR |
5–200 |
0.9996 |
1 |
5 |
4.5 |
| LUT |
1–200 |
0.9989 |
0.5 |
1 |
4.3 |
| KAE |
1–200 |
0.9979 |
0.5 |
1 |
2.4 |
| API |
5–200 |
0.9973 |
1 |
5 |
2.8 |
3.4 Application to real samples
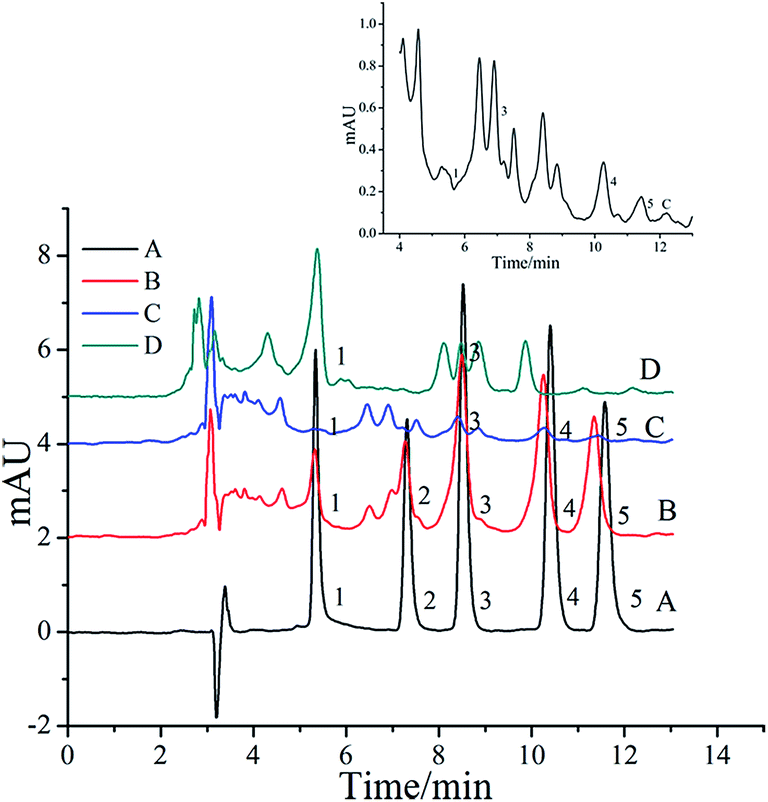
Flavonoids, as secondary metabolites, widely exist in grapes and a large number of other natural plants.29 In order to certify the reliability of the proposed SPE-HPLC-DAD method, two different grape juice drink samples, bought from a supermarket as real samples, were analyzed by HPLC-DAD combined with SPE under the optimal conditions described above. The chromatograms obtained from the two real samples and standard solution are shown in Fig. 10. The analytes of MYR, LUT, KAE, and API in grape juice sample 1# were detected, and MYR was quantified at 18.1 ng mL−1. The analytes of MYR and LUT in the grape juice sample 2# were detected, and MYR was quantified at 72.6 ng mL−1. Recovery was studied with an addition level at 20 ng mL−1 for all the selected flavonoids in sample 1#, and the results are shown in Table 2. The recoveries of all the selected flavonoids were in the range of 91.5–120.7%. Therefore, the method was reliable enough for practical applications.
 |
| | Fig. 10 Chromatograms of MYR (1), QUR (2), LUT (3), KAE (4) and API (5) in grape juice drinks. A: concentration of 1 ng mL−1 of standard solution. B: spiked with 20 ng mL−1 of grape juice sample 1# by proposed SPE method. C: grape juice sample 1# by proposed SPE method. D: grape juice sample 2# by proposed SPE method. | |
Table 2 Determination results and recovery of flavonoids in the grape juice drinks and recovery based on five replicates
| Flavonoids |
Flavonoids in the grape juice drinking (ng mL−1) |
Recoverya for standard addition (n = 5, %) |
| Sample 1# |
Sample 2# |
| Spiked level for all flavonoids: 20 ng mL−1. RSD% based on three replicates in the real sample. |
| MYR |
18.1 ± 2.2b |
72.6 ± 4.3b |
91.5 ± 9.1 |
| QUR |
Not detected |
Not detected |
91.6 ± 5.9 |
| LUT |
Detected but not quantified |
Detected but not quantified |
95.6 ± 3.7 |
| KAE |
Detected but not quantified |
Not detected |
97.0 ± 4.4 |
| API |
Detected but not quantified |
Not detected |
120.7 ± 5.9 |
4. Conclusions
In this work, a novel material was prepared by introducing Nbim into pure silica and was successfully used for determination of flavonoids coupled with HPLC-DAD. The extraction efficiency of the Nibm-modified silica sorbent was significantly higher than that of the diol-modified silica sorbent. The satisfactory extraction efficiency and good extraction capability are mainly attributed to multiple intermolecular interactions, including π–π stack, hydrophobic, and hydrogen bonding interactions between the Nbim-modified silica sorbent and flavonoids. Under the optimal conditions, modified silica was used as the SPE sorbent for extraction of flavonoids from grape juice drinks and the new sorbent showed acceptable sensitivity and extraction efficiency, indicating that the new Nibm-modified silica could serve as a promising sorbent for the SPE of polyphenolic compounds from complex matrices with sufficient sensitivity and accuracy. This method will provide a powerful way to detect polyphenolic compounds and quantify them in complex matrices, as well as to assess the quality of some drinks.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (20972170 and 21275150) and the Funds for Distinguished Young Scientists of Gansu (1210RJDA013).
References
- S. Cicerale, L. Lucas and R. Keast, Int. J. Mol. Sci., 2010, 11, 458–479 CrossRef CAS PubMed.
- S. H. Omar, Sci. Pharm., 2010, 78, 133–154 CrossRef CAS PubMed.
- M. Servili, S. Esposto, R. Fabiani, S. Urbani, A. Taticchi, F. Mariucci, R. Selvaggini and G. F. Montedoro, Inflammopharmacology, 2009, 76–84 CrossRef CAS PubMed.
- M. López, F. Martínez, C. Del Valle, M. Ferrit and R. Luque, Talanta, 2003, 60, 609–616 CrossRef.
- F. Fang, J. M. Li, Q. H. Pan and W. D. Huang, Food Chem., 2007, 101, 428–433 CrossRef CAS PubMed.
- I. Klimczak, M. Małecka, M. Szlachta and A. Gliszczyńska-Świgło, J. Food Compos. Anal., 2007, 20, 313–322 CrossRef CAS PubMed.
- M. N. Irakli, V. F. Samanidou, C. G. Biliaderis and I. N. Papadoyannis, J. Sep. Sci., 2012, 35, 1603–1611 CrossRef CAS PubMed.
- J. V. Formica and W. Regelson, Food Chem. Toxicol., 1995, 33, 1061–1108 CrossRef CAS.
- P. Knekt, R. Järvinen, R. Seppänen, M. Heliövaara, L. Teppo, E. Pukkala and A. Aromaa, Am. J. Epidemiol., 1997, 146, 223–230 CrossRef CAS.
- G. Theodoridis, M. Lasakova, V. Skerikova, A. Tegou, N. Giantsiou and P. Jandera, J. Sep. Sci., 2006, 29, 2310–2321 CrossRef CAS.
- E. Tahmasebi and Y. Yamini, Anal. Chim. Acta, 2012, 756, 13–22 CrossRef CAS PubMed.
- S. F. Xu, L. X. Chen, J. H. Li, W. Qin and J. P. Ma, J. Mater. Chem., 2011, 21, 12047–12053 RSC.
- J. P. Ma, R. H. Mao, J. H. Li, X. H. Zhao, B. Z. Shi and S. Q. Li, J. Chromatogr. Sci., 2009, 47, 110–115 CAS.
- Y. H. Chen, T. T. Wang, J. F. Ma, Z. Liang, M. L. Chen, J. H. Fang, H. Q. Gao, L. H. Zhang and Y. K. Zhang, J. Sep. Sci., 2014, 37, 143–150 CrossRef CAS PubMed.
- E. Tahmasebi, Y. Yamini, A. Mehdinia and F. Rouhi, J. Sep. Sci., 2012, 35, 2256–2265 CrossRef CAS PubMed.
- Y. P. Li, X. Li and Z. Zhou, RSC Adv., 2014, 4, 39192–39196 RSC.
- M. R. Khan, M. A. Khan, Z. A. Alothman, I. H. Alsohaimi, M. Naushad and N. H. Al-Shaalan, RSC Adv., 2014, 4, 34037–34044 RSC.
- X. Y. Lai, Y. Y. Zhao, H. Liang, Y. J. Bai, B. Wang and D. Guo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 852, 108–114 CrossRef CAS PubMed.
- Y. Zhang, B. L. Bao, B. Y. Lu, Y. P. Ren, X. W. Tie and Y. Zhang, J. Chromatogr. A, 2005, 1065, 177–185 CrossRef CAS PubMed.
- M. S. Dopico-Garcia, P. Valentao, L. Guerra, P. B. Andrade and R. M. Seabra, Anal. Chim. Acta, 2007, 583, 15–22 CrossRef CAS PubMed.
- L. S. Qing, J. Xiong, Y. Xue, Y. M. Liu, B. Guang, L. S. Ding and X. Liao, J. Sep. Sci., 2011, 34, 3240–3245 CrossRef CAS PubMed.
- L. T. Wang, W. Wei, Y. Guo, J. Xu and S. J. Shao, Spectrochim. Acta, Part A, 2011, 78, 726–731 CrossRef PubMed.
- M. Sun, H. D. Qiu, L. C. Wang, X. Liu and S. X. Jiang, J. Chromatogr. A, 2009, 1216, 3904–3909 CrossRef CAS PubMed.
- Z. Jiang, J. W. Shao, M. L. Chen, J. Wang and L. Jia, Analyst, 2013, 138, 4385–4392 RSC.
- T. T. Wang, Y. H. Chen, J. F. Ma, M. L. Chen, C. G. Nie, M. J. Hu, Y. Li, Z. J. Jia, J. H. Fang and H. Q. Gao, J. Chromatogr. A, 2013, 1308, 63–72 CrossRef CAS PubMed.
- C. L. Silva, J. L. Goncalves and J. S. Camara, Anal. Chim. Acta, 2012, 739, 89–98 CrossRef CAS PubMed.
- J. P. Ma, R. H. Xiao, J. H. Li, J. B. Yu, Y. Q. Zhang and L. X. Chen, J. Chromatogr. A, 2010, 1217, 5462–5469 CrossRef CAS PubMed.
- J. J. Feng, M. Sun, L. L. Xu, J. B. Li, X. Liu and S. X. Jiang, J. Chromatogr. A, 2011, 1218, 7758–7764 CrossRef CAS PubMed.
- F. S. Mirnaghi, F. Mousavi, S. M. Rocha and J. Pawliszyn, J. Chromatogr. A, 2013, 1276, 12–19 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13861a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.