
Electronic and structural properties of bulk arsenopyrite and its cleavage surfaces – a DFT study†
Juliana C. M. Silva,
Heitor A. De Abreu and
Hélio A. Duarte*
Grupo de Pesquisa em Química Inorgânica Teórica – GPQIT – Departamento de Química, ICEx, Universidade Federal de Minas Gerais (UFMG), Av. Antônio Carlos, 6627, 31270-901 Belo Horizonte, MG, Brazil. E-mail: duarteh@ufmg.br
First published on 13th November 2014
Abstract
Arsenopyrite is the most abundant arsenic containing mineral on Earth and it is normally associated with many other minerals of economic importance. Therefore, it is involved in the environmental impacts of mining activities. The bonding nature of arsenopyrite and its preferential cleavage surface are still controversial. In the present work we have investigated the structural and electronic properties of arsenopyrite and its cleavage surface formation using a density functional/plane waves method. The quantum theory of atoms in molecules (QTAIM) and electron localization function (ELF) were applied for investigating the nature of the bonding in arsenopyrite. No evidence was found for Fe–Fe bonding in the bulk structure. The As–S bond has large covalent character and it is unexpected to be broken in the surface formation. The cleavage and surface energies have been calculated indicating that the (001) surface is the most favored.
1. Introduction
Arsenopyrite is a mineral sulfide commonly found in nature and is associated with noble metals such as gold, copper and silver. This mineral has little economic value1 and the mining process generates a huge quantity of tailings which are disposed in dams.2 The exposition of sulfide minerals, such as arsenopyrite, to the environment normally leads to acid rock drainage (ARD).During ARD, the mineral sulfides present in the mining tailings oxidize producing sulfuric acid. The generated solution acts as a leaching agent, i.e., as a mixture that solubilizes the solid mineral constituents, producing an acidic solution containing dissolved metals, which can contaminate soil and underground water. ARD is a spontaneous process that arises where the rock is exposed to air and moisture. A classic example occurs in Río Tinto in Spain.3 Along this river there is a large deposit of pyrite and the process of ARD has occurred for centuries, leading to a pH of about 2 with a high concentration of heavy metals. However, ARD normally occurs due to anthropogenic activities, such as mining, which expose large amounts of sulfide minerals to the atmosphere.4
Although arsenopyrite is stable under reducing conditions, its oxidation due to weathering effects releases sulfate, arsenite (As(III)) and arsenate (As(V)) species1 to the environment. The arsenic released due to arsenopyrite oxidation is an environmental hazard and may become a health problem.5 The understanding of the kinetics and the mechanism of dissolution of this material in different conditions is essential for assessing the stability of the arsenic containing tailings and the development of more efficient processes to control its remobilization with great environmental, social and economic consequences.
There are several studies in the literature concerning the products formed in arsenopyrite oxidation in different media.1,2,6–11 However, in many of them the results diverge and there is no consensus concerning the reaction mechanism at a molecular level. In this context, first-principles calculations emerge as a tool for providing information about the surface reactivity of arsenopyrite and insights about its oxidation mechanism. For pyrite there are many theoretical studies concerning its cleavage surfaces,12,13 water adsorption14,15 and mechanism of oxidation,16 as well as about chalcopyrite’s surfaces and adsorption of leaching agents.17–20 Nevertheless, to the best of our knowledge, there is only one theoretical study about the electronic and geometric characteristics of the (110) surface of arsenopyrite21 and two studies about arsenic incorporation into pyrite.22,23 More information about arsenopyrite and its chemical reactivity is therefore necessary.
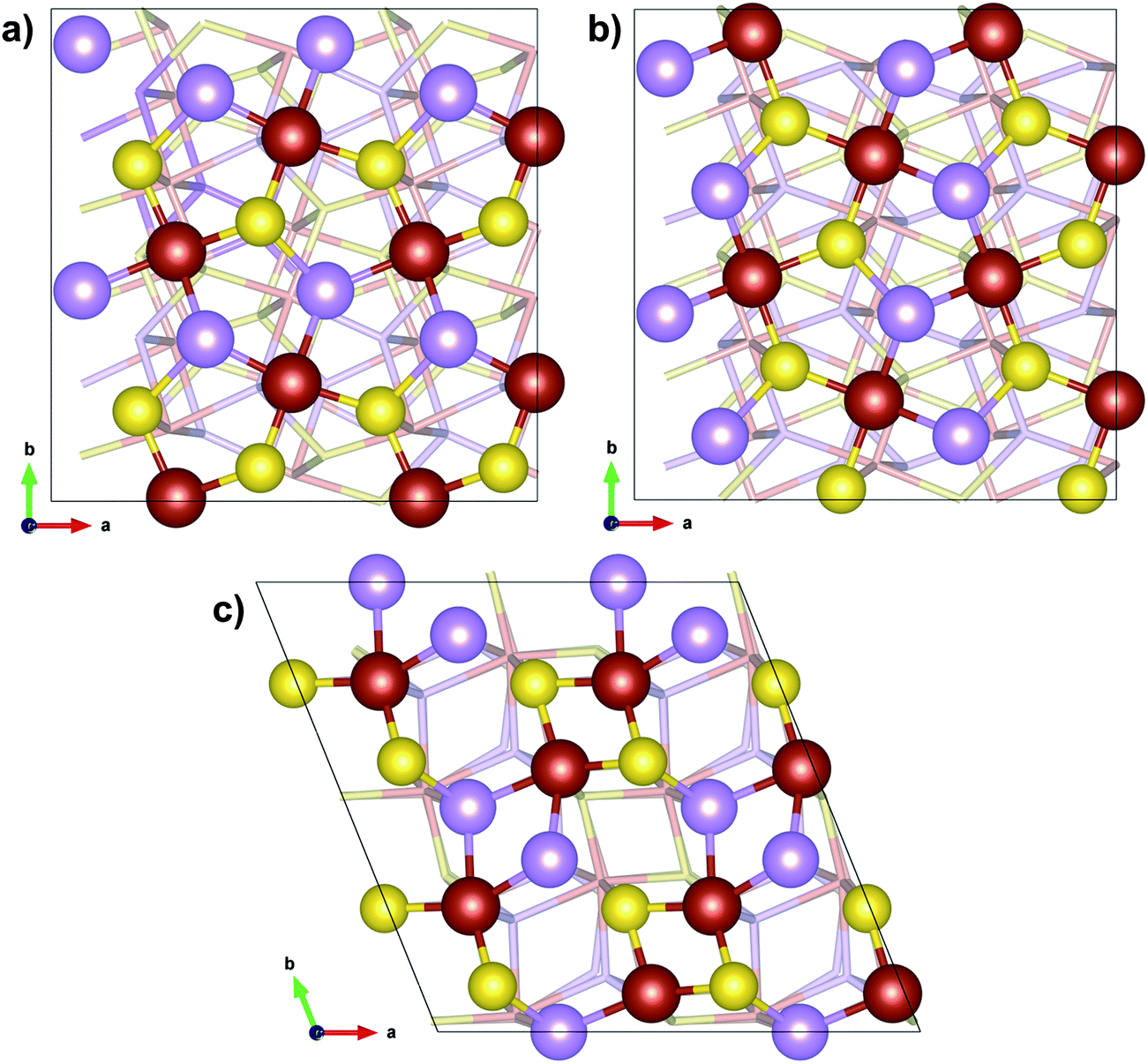
Arsenopyrite (ideal formula FeAsS) is the most common arsenic containing mineral on Earth and may be found in many ore deposits. It is a diamagnetic semiconductor.24 Its unit cell is monoclinic and has a space group P21/c derived from marcasite (orthorhombic FeS2)25 with 4 FeAsS per unit cell, as shown in Fig. 1a and b. However, a refinement of the arsenopyrite structure was also accomplished in space group C21/d,26 in a pseudo-orthorhombic unit cell, as shown in Fig. 1c. Its structure contains arsenic and sulfur dianions (As–S) coordinated to the iron atom in an octahedral shape (Fig. 2a). The Fe atoms are coordinated to three As and three S atoms and each anion is coordinated to three iron atoms and another anion in a tetrahedral shape. The FeAs3S3 octahedrons are asymmetric and share two opposite edges to form single strips parallel to the (1![[0 with combining macron]](https://www.rsc.org/images/entities/char_0030_0304.gif) 1) surface (Fig. 2b). The adjacent octahedral coordinations in a row are related to each other by an inversion operation, leading to distances between Fe cations alternating between short and long. In the direction of the Fe–Fe contact, the short Fe–Fe bond cuts the long S–S edge, whereas the long Fe–Fe bond cuts the short As–As edge. Natural arsenopyrite has a composition ranging from FeAs0.9S1.1 to FeAs1.1S0.9
1) surface (Fig. 2b). The adjacent octahedral coordinations in a row are related to each other by an inversion operation, leading to distances between Fe cations alternating between short and long. In the direction of the Fe–Fe contact, the short Fe–Fe bond cuts the long S–S edge, whereas the long Fe–Fe bond cuts the short As–As edge. Natural arsenopyrite has a composition ranging from FeAs0.9S1.1 to FeAs1.1S0.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and there may be metal impurities such as Co replacing Fe26 in the structure. Mössbauer spectroscopy indicates that the Fe atom is divalent in the low-spin state, in an octahedral environment.28,29
27 and there may be metal impurities such as Co replacing Fe26 in the structure. Mössbauer spectroscopy indicates that the Fe atom is divalent in the low-spin state, in an octahedral environment.28,29
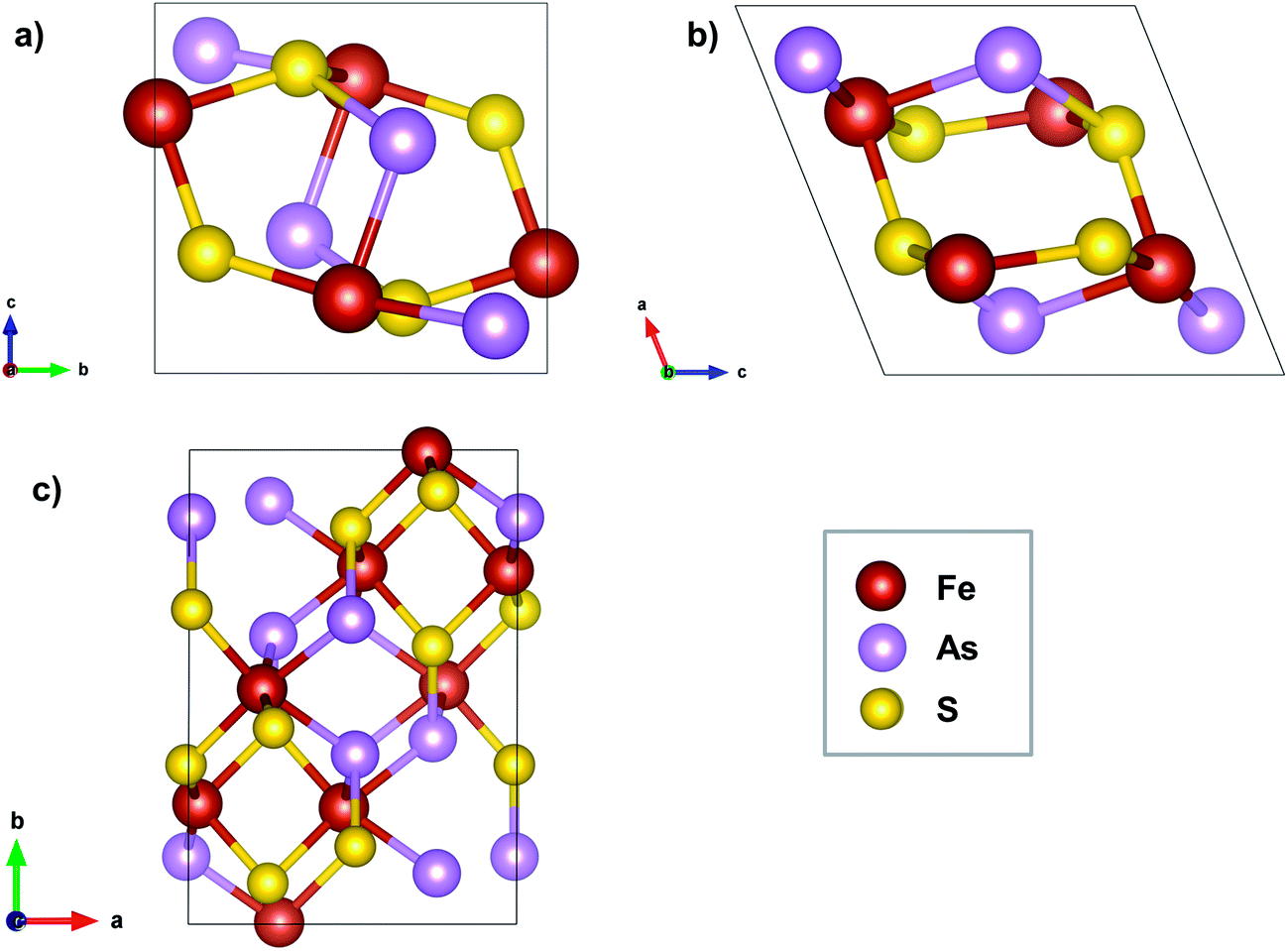 | ||
| Fig. 1 Arsenopyrite structure. (a) View of the monoclinic cell along the a axis; (b) view of the monoclinic cell along the b axis; (c) view of the pseudo-orthorhombic cell along the c axis. | ||
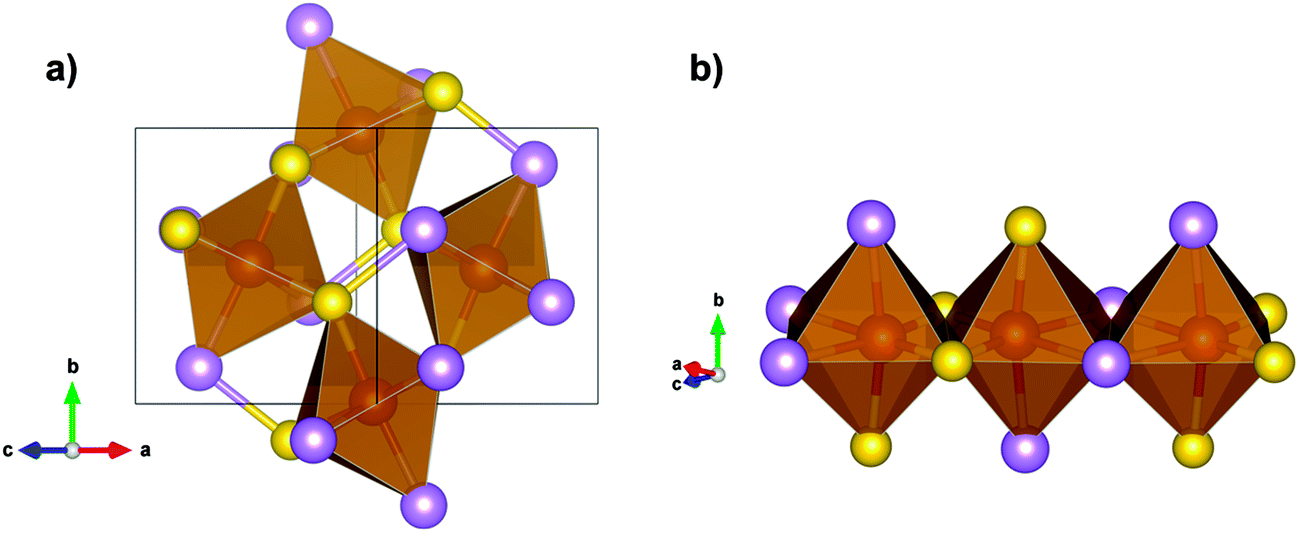 | ||
| Fig. 2 (a) As–S dianions octahedrally coordinated to Fe. (b) Neighboring octahedrons sharing one edge. Yellow atoms are sulfur, purple are arsenic and red are iron. | ||
Concerning the favorable cleavage plane, there is no consensus regarding which is the favored plane among the (100),11 (001),30 (101)25,31 and (110)21,32 planes. Many of the reported works did not define clearly which is the unit cell used as the reference, leading to some ambiguity in defining the favored cleavage.
In the present work, the structural and electronic properties of the arsenopyrite bulk and different cleavage surfaces have been investigated aiming to fulfil the lack of information about this system.
2. Methodology
The calculations have been performed based on the density functional theory (DFT)/plane waves methodology with periodic boundary conditions as implemented in the Quantum Espresso package.33 The PW9134 exchange/correlation (XC) functional and ultrasoft pseudopotentials proposed by Vanderbilt35 with the following valence configurations: Fe (3s2 3p6 3d6.5 4s1 4p0), As (4s2 4p3) and S (3s2 3p4) were used. For the bulk, a cutoff energy of 60 Ry was used and a 4 × 4 × 4 K-point mesh sampling based on the Monkhorst-Pack scheme36 was chosen, besides Marzari–Vanderbilt37 0.02 Ry smearing. For the surface, 30 Ry energy cutoff and a 2 × 2 × 1 K-point mesh was used. The energy was converged to 10−9 Ry. All surfaces were set using a (2 × 2 × 2) slab model with 15 Å of vacuum.All calculations were spin compensated. Geometry optimization was carried out using a damped dynamics method38 with a Parrinello-Rahman extended Lagrangian,39 keeping a force tolerance criterion of 10−3 Ry Bohr−1. The atomic positions, as well as the cell parameters, were fully optimized for the bulk. For the slab calculations only the atomic positions were fully optimized.
Bader’s QTAIM (Quantum Theory of Atoms In Molecules) method40 was used to investigate the electronic structure of the solid using the program Critic2.41 For the bulk modulus calculation, the program Gibbs242 was used. Band structure, density of states (DOS), electron localization function (ELF) and electron density plots were built using the Quantum Espresso code and a 8 × 8 × 8 mesh of K-points and for the QTAIM analysis the electron density was built using a 12 × 12 × 12 mesh of K-points.
3. Results and discussion
Bulk
The calculated interatomic distances of arsenopyrite bulk are shown in Table 1 and compared with available experimental values. The theoretical estimates are closer to the experimental results of Bindi et al.29 with a maximum of 2% difference, which are more recent and refer to a sample rich in As. The average Fe–S distance is 2.203 Å, 0.028 Å less than the experimental value29 and also less than the values calculated using similar methodologies for marcasite (orthorhombic FeS2, 2.23 Å)43 and chalcopyrite (CuFeS2, 2.241 Å),17 as expected. The average Fe–As distance of 2.402 Å is 0.005 Å larger than the experimental value,29 and the As–S distance of 2.405 Å is 0.031 Å larger. The As–S bond length is also larger than the S–S distance calculated by Gudelli et al.43 for marcasite (2.20 Å), which is expected due to the larger atomic radius of As. Gudelli et al.43 also found only one Fe–Fe distance for marcasite (3.38 Å), an intermediate value between both distances found in the present work.| Reference | Short Fe–Fe | Long Fe–Fe | Fe–S | Fe–As | As–S |
|---|---|---|---|---|---|
| This work | 2.668 | 3.765 | 2.190 | 2.380 | |
| 2.198 | 2.410 | 2.405 | |||
| 2.222 | 2.415 | ||||
| Experimental (1961)27 | 2.82 | 3.62 | 2.22, 2.24 | 2.30, 2.32 | |
| 2.25, 2.26 | 2.32, 2.38 | 2.33 | |||
| 2.26, 2.29 | 2.39, 2.41 | ||||
| Experimental (1987)26 | 2.922 | 3.627 | 2.239 | 2.336 | |
| 2.250 | 2.371 | 2.346 | |||
| 2.257 | 2.375 | ||||
| Experimental (2012)29 | 2.734 | 3.741 | 2.229 | 2.370 | |
| 2.230 | 2.409 | 2.374 | |||
| 2.233 | 2.412 |
The cell parameter results after optimization are shown in Table 2. They are in agreement with the experimental data and are closer to these data than the DFT/PBE/plane waves results of Corkhill et al.21 The differences between lattice parameters did not exceed 0.022 Å, 0.33 degrees in β and 1.72 Å3 in volume, compared to Bindi et al.29
Several studies tried to explain the differences in pyrite (cubic FeS2), marcasite (orthorhombic FeS2), arsenopyrite (FeAsS) and lollingite (FeAs2) structures. Hulliger and Mooser,44 Pearson45 and Nickel46,47 used ligand field theory to explain the stabilities of each structure. In pyrite, neighboring octahedrons share common corners, while in the other three sulfides, they share edges. In lollingite, the Fe–Fe distance is shorter than in marcasite, and in arsenopyrite there are alternating short and long distances. In these works, as well as in the works of Brostigen and Kjekshus,48 and Goodenough,49 the iron atom in arsenopyrite was considered to be in the trivalent oxidation state. These authors, except Goodenough,49 considered that there should be an Fe–Fe bond in arsenopyrite to explain the fact that iron is trivalent, but in a low spin state in this mineral, since arsenopyrite is diamagnetic. In fact Pauling50 has found complexes with an Fe–Fe bond up to 2.78 Å long. Vaughan and Craig,28 and Bindi et al.29 showed through Mössbauer spectroscopy that iron is actually in a divalent state for all of these sulfides. Tossell et al.51 considered this result and calculated the ionization energy of orbitals in a MA6 model for sulfides through the SCF-Xα-SW method. They concluded that these structures are defined by a metal–dianion interaction and proposed an explanation for the different structures based on molecular orbital occupation. Schmokel and coworkers52 made a detailed analysis of the experimental and theoretical electron density of pyrite and marcasite. They found that S–S bonds are more covalent and Fe–S bonds are weaker in pyrite when compared to marcasite. This is explained based on the distribution of the d-orbital-like density difference between the two polymorphs.
XPS experiments performed by Nesbitt et al.1,53 checked that in bulk arsenopyrite and lollingite, the iron is divalent because the main peak on the spectra is very close in bonding energy to pyrite’s spectra. They also suggested that As and S atoms are in the (−1) oxidation state in arsenopyrite. Then, it is accepted that the oxidation states in arsenopyrite are Fe2+As−S−.
In order to better understand the electronic properties of arsenopyrite and also validate the bulk model, the band structure was calculated using the K-points path suggested by Setyawan et al.54 for a monoclinic cell, see Fig. S1.† It can be seen by the band structure shown in Fig. 3 that arsenopyrite is a semiconductor, according to the literature.55 The indirect band gap of 0.75 eV from the D to Γ point is shown with the red vector. This value is 0.07 eV lower than the experimental value of 0.82 eV,9 as expected, since it is well known that the GGA XC functional underestimates the band gap.56 The band gap values for pyrite vary from 0.7 to 2.62 eV, but the most reliable ones are between 0.9 and 0.95 eV, measured in photoconductivity experiments.57,58 Opahle et al.59 found a value of 0.85 eV in a DFT/LDA-PZ calculation for pyrite, the same difference from experimental values was found for arsenopyrite in the present work. Gudelli et al.43 calculated a band gap of 1.186 eV for pyrite and 1.603 eV for marcasite using the TBmBJ potential,56 which are overestimated.
 | ||
| Fig. 3 Band structure calculated for arsenopyrite. | ||
The total and projected density of states (DOS) are shown in Fig. 4. The iron atom is the one that most contributes to the states around the Fermi level, in both the valence and conduction bands, especially the d orbitals of iron (Fig. S2†). According to the discussion made by De Oliveira and Duarte,17 this characteristic is coherent with an Fe atom in the oxidation state (II) since this atom can be oxidized as well as reduced.
 | ||
| Fig. 4 Total and projected DOS over the atoms of arsenopyrite plotted using a Gaussian width of 0.005 Ry. | ||
The electron density map of the (010) plane is shown on Fig. 5, indicating that the electron density between neighboring Fe atoms is very small.
 | ||
| Fig. 5 Electronic density map of the arsenopyrite (010) plane. Red balls are iron atoms. | ||
QTAIM analysis was performed for arsenopyrite in order to characterize the chemical bonding in the structure. A total of 90 critical points were found, with 24 of them being nonequivalent: 3 nuclei critical points (NCP), 7 bond critical points (BCP), 9 ring critical points (RCP) and 5 cage critical points (CCP). Their positions are shown in Fig. 6 and the respective data are shown on Table 3 (a complete table is shown as Table S2†). The Morse relationship ensures that n − b + r − c = 0, with n, c ≥ 1, and b, r ≥ 3 which holds for the nonequivalent points.
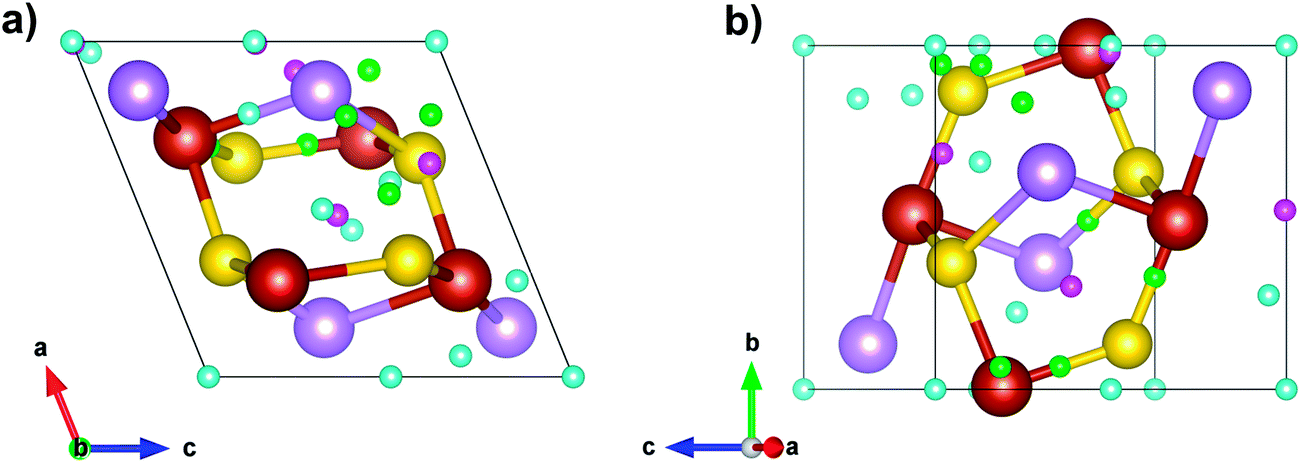 | ||
| Fig. 6 Bond critical points (BCP) in green, ring critical points (RCP) in blue, and cage critical points (CCP) in pink for arsenopyrite QTAIM analysis. Atoms in red are iron, in yellow are sulfur and in purple are arsenic. | ||
| Critical points | ρ(rc)a | ∇2ρ(rc)a | Chemical meaning |
|---|---|---|---|
| a In atomic units. | |||
| Fe | 6.2235 | −73.0292 | |
| As | 0.5412 | −18.0058 | |
| S | 0.3588 | −10.8180 | |
| b1 | 0.0721 | 0.0492 | Fe–As |
| b2 | 0.0961 | 0.1501 | Fe–S |
| b3 | 0.0733 | 0.0730 | Fe–As |
| b4 | 0.0924 | 0.1996 | Fe–S |
| b5 | 0.0725 | 0.0475 | Fe–As |
| b6 | 0.0828 | −0.0075 | As–S |
| b7 | 0.0881 | 0.2232 | Fe–S |
| r1 | 0.0326 | 0.0546 | Fe⋯Fe long |
| r2 | 0.0430 | 0.0539 | Fe⋯Fe short |
Concerning the BCPs, three are Fe–S bonds, three are Fe–As bonds and one is an As–S bond, as shown in Fig. 7a. The critical points involving Fe–S bonds have a positive Laplacian indicating a larger ionic character. Schmokel et al.52 have proposed that the Fe–S bond in pyrite and marcasite has some covalent character due to Fe d orbitals mixing involved in this bond. The Fe–S BCP shows values of density and Laplacian (see Table 3) close to those of Gibbs et al.60 calculated using DFT/LDA and localized basis sets, and also close to the values published by Schmokel et al.52 in a DFT/PBE study using multipole refinement. Aray et al.61 found a value of 0.079 a.u. of density for the Fe–S BCP in pyrite and 0.13251 a.u. for the S–S BCP in DFT/PBE (FP-LAPW, full-potential linearized augmented plane-wave + local orbitals) calculations. The Fe–As BCP has a density and positive Laplacian smaller than the Fe–S BCP indicating that Fe–S has larger ionic character and is stronger than the Fe–As bonding. The As–S BCP has a density of 0.0828 a.u. and Laplacian of −0.0075, which means it has covalent character. Schmokel et al.52 found similar results for density and Laplacian of S–S BCP in pyrite and marcasite. The electron density and Laplacian of the S–S BCP in marcasite is 0.115 a.u. and −0.015 a.u., respectively, compared to the values of the same bond in pyrite of 0.126 a.u. and −0.043 a.u., respectively, indicating that this bond is stronger in pyrite than in marcasite. This result is similar to the density of S–S BCP in covellite (0.135 a.u.) and its Laplacian (−0.079 a.u.) found by Morales-García et al.62 using a similar computational method. Comparing with our results for As–S BCP in arsenopyrite of 0.083 and −0.007 a.u. for density and Laplacian, respectively, it is expected that the As–S bond in arsenopyrite is weaker than the S–S bond in pyrite, marcasite and covellite. However, analyzing the values shown in Table 3, the As–S bond is strongest in arsenopyrite and, therefore, unlikely to break in surface cleavage.
 | ||
| Fig. 7 (a) Bond critical points and (b) ring critical points in detail. Yellow atoms are sulfur, purple are arsenic and red are iron. | ||
The ELF (electron localization function) map was generated (Fig. 8) for the As–S bond. In the region where the ELF value is close to 1, the electrons are localized, and in the regions where they are delocalized, like a homogeneous electron gas, as in metallic bonding, it has a value of 0.5. In Fig. 8, it is possible to see the shared cloud between As and S atoms, indicating a covalent bond.
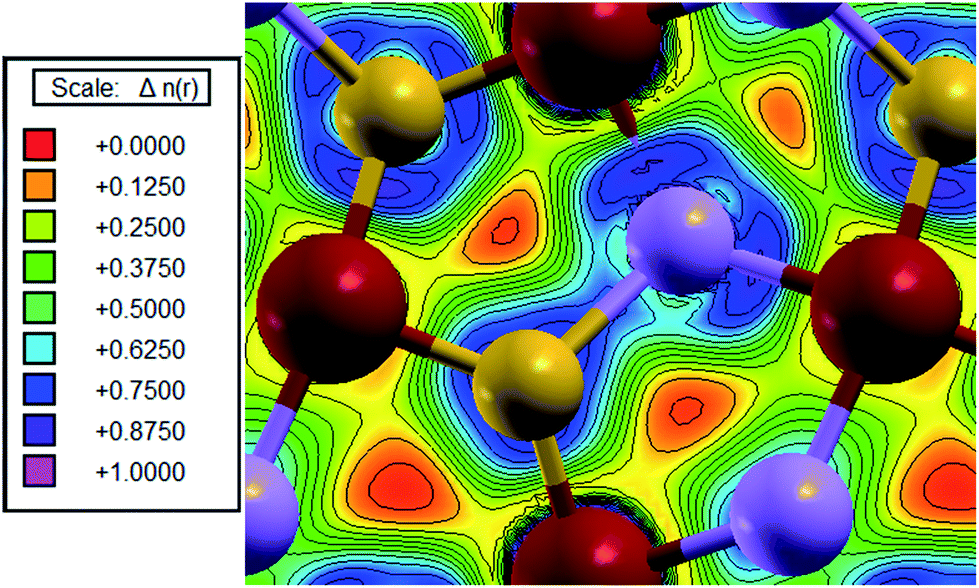 | ||
| Fig. 8 ELF of the arsenopyrite As–S bond. Red is iron, purple is arsenic and yellow is sulfur. | ||
In the QTAIM analysis no evidence of the Fe–Fe bond has been found in arsenopyrite. A ring critical point (RCP) between two neighboring iron atoms was found as expected, see Fig. 7b. From our QTAIM analysis, the hypothesis of the Fe–Fe bond is completely discharged. The values of density and Laplacian for the r1 and r2 RCPs corresponding to the Fe⋯Fe distances of 3.765 and 2.668 Å are very similar and the interaction of these atoms must have the same nature. The ELF map was generated (Fig. 9) for the Fe–Fe region. The ELF value between two Fe atoms is estimated to be 0.25, which is not characteristic of chemical bonding.
 | ||
| Fig. 9 ELF of the arsenopyrite (010) plane. Red balls are iron. | ||
The calculated atomic charges and volumes in QTAIM are reported in Table 4. The estimated positive atomic charge for As is reasonable since sulfur is more electronegative. The integration of the AsS basins leads to −0.41e and 0.41e for the iron basin. This is in agreement with Fe2+(AsS)2−. The As and S atoms have the largest volumes, which means that these atoms dominate the crystal compressibility.
| Atoms | Charge | Volume (a.u.) | Pauling’s electronegativity |
|---|---|---|---|
| Fe | 0.41 | 69.9 | 1.83 |
| As | 0.18 | 112.9 | 2.18 |
| S | −0.59 | 111.8 | 2.58 |
| Total | 1178.5 |
The c parameter63 can be used to evaluate the difference between topological charge, Q(Ω), and nominal oxidation state, OS(Ω), according to eqn (1) and it indicates the ionicity of the crystal.
 | (1) |
The closer to 1 c is, the more ionic the crystal, and the closer to 0, the more covalent it is. The value of c for arsenopyrite is 0.205 taking Fe2+As1−S1− oxidation numbers as reference, which indicates a solid that presents large covalent character.
The bulk modulus of arsenopyrite was calculated and compared with experimental and theoretical values of other compounds, as shown in Table 5. Our value of 147.5 GPa is 15 GPa greater than the experimental value obtained by Fan et al.64 in X-ray diffraction in the pressure range from 0 to 9.6 GPa. The calculated and experimental values for marcasite and pyrite are in the same level of magnitude as our results. This shows that the method used in this work describes well not just each atom individually, but also the whole crystal structure.
Surface
To the best of our knowledge, there is no consensus in the literature concerning the favored arsenopyrite cleavage. The (100),11 (001),30 (101)25,31 and (110)21,32 surfaces have been indicated by different references as most favorable to be cleaved. It is important to highlight that in some of these works the unit cell used to define the Miller indices was not clearly indicated explaining part of this divergence. Table S1† shows the correspondence between the C21/d and P21/c unit cells which are normally used to describe arsenopyrite. We stress that it is important to clearly define the unit cell used to define the surfaces.Several surfaces were created based on the P21/c optimized bulk cell to compare their energies and the planes are shown in Fig. S3.† The non-optimized structures are presented in Fig. S4.† All the different terminations were calculated. All of them are type II from Tasker,68 namely, they had charged layers formed by cations and anions symmetrically arranged in a way that the charges cancel on the surface and they do not have a resultant dipole perpendicular to the surface. Usually, this means that there is no reconstruction of these surfaces, only relaxation. The surface energy was calculated according to eqn (2):69
 | (2) |
| Surface | Surface energy/J m−2 | Cleavage energy/J m−2 | Coordination | ||
|---|---|---|---|---|---|
| Fe | As | S | |||
| Bulk | — | — | 6 | 4 | 4 |
| 001 | 1.05 | 1.23 | 5 | 3 | 3 |
| 010 | 1.06 | 1.28 | 5 | 3 | 3 |
| 100 As-terminal | 1.07 | 1.21 | 5 | 3 | 4 |
| 100 S-terminal | 1.09 | 1.35 | 5 | 4 | 4 |
| 011 | 1.30 | 1.50 | 5, 4 | 2 | 3 |
| 101 | 1.47 | 1.65 | 4 | 3 | 3 |
| 110 S-terminal | 1.52 | 1.91 | 4 | 3 | 3, 2 |
| 110 As-terminal | 1.57 | 1.93 | 4 | 3, 2 | 3 |
| 111 | 1.51 | 1.78 | 5 | 2 | 2 |
| 210 (1) | 1.44 | 1.59 | 4, 3 | 3, 2 | 4, 3 |
| 210 (2) | 1.78 | 2.29 | 4, 3 | 3, 1 | 2 |
 | ||
| Fig. 10 Optimized arsenopyrite surfaces. Iron is in red, sulfur in yellow and arsenic in purple. Miller indices are based on the P21/c symmetry. | ||
As one can see in Table 6, the surface and cleavage energies are related to the number of bonds broken. The (001), (010), and (100) planes are the most favored cleavage surfaces with Esurf between 1.05 and 1.09 J m−2 and Ecleav between 1.21 and 1.35 J m−2. The (100) plane is more likely to cleave on the As-terminal surface. The top view of these planes is presented in Fig. 11, which shows that the (001) and (100) surfaces expose a similar terminal structure. In these planes no As–S bond is broken, as expected. The other ones have higher energies and are unlikely to be cleaved. Nevertheless, they may be exposed when fractured due to twinning along the high cleavage energy planes. According to Klein et al.,25 twinning appears on surfaces (100) and (001) of the P21/c cell, whereas according to Dana and Ford32 it appears on surfaces (110) and (101).
 | ||
| Fig. 11 Top view of surfaces: (a) (001); (b) (100); (c) (010). Red is iron, purple is arsenic and yellow is sulfur. | ||
Pyrite has a cubic unit cell, so the (100), (010), and (001) surfaces are equivalent and have been used in adsorption or oxidation studies of pyrite.14,16 Hung et al.12,13 have studied pyrite’s (100), (110), (111), and (210) surfaces through PBE/plane waves calculations. Their results follow the same tendency of arsenopyrite, except for the high value in (100) cleavage energy of 4.25 J m−2. Table 7 shows a comparison between the surface and cleavage energies for arsenopyrite and pyrite. However, it is important to note that pyrite and arsenopyrite present different unit cells, therefore the bond breaking in the surface formation is different.
| Surface | Arsenopyrite | Pyrite12,13 | ||
|---|---|---|---|---|
| Surface energy/J m−2 | Cleavage energy/J m−2 | Surface energy/J m−2 | Cleavage energy/J m−2 | |
| a Microfacetted (110) pyrite surface.b Corkhill et al.21c The (001), (010) and (100) pyrite surfaces are equivalent. | ||||
| 001 | 1.05 | 1.23 | 1.06c | 4.25c |
| 010 | 1.06 | 1.28 | ||
| 100 | 1.07 | 1.21 | ||
| 110 (S-terminal) | 1.52 | 1.91 | 1.68 | 1.85 |
| 110 (As-terminal) | 1.57 | 1.93 | 1.54a | 1.74a |
| 110 (1)b | 1.76 | 2.08 | — | — |
| 110 (2)b | 2.08 | 2.40 | — | — |
| 111 | 1.51 | 1.78 | 1.40 | 1.61 |
| 210 | 1.44 | 1.59 | 1.50 | 1.74 |
Corkhill et al.21 also calculated the Esurf and Ecleav of (110) arsenopyrite surfaces in two different terminations using PBE/plane waves. The models used by them (24 atoms, 5.65 Å × 7.91 Å × 16.9 Å cell) are four times smaller than the ones considered in the present work and do not have equal terminations on the top and bottom surfaces. Their estimated values of Esurf (1.76 and 2.08 J m−2), and Ecleav (2.08 and 2.40 J m−2) are higher than our results, with both presented in Table 7.
The largest change in the relaxed slab model was observed for the surface atoms with values of 0.12 Å for Fe–As bond on the (100) surface, 0.14 Å for Fe–As bond on the (001) surface, and 0.14 Å for Fe–Fe distance on the (010) surface.
The projected DOS over the surface atoms of the (001), (010), and (100) planes are shown in Fig. 12. Similarly to the bulk, the Fe 3d orbitals are dominant around the Fermi level, which means that nucleophilic or electrophilic interactions with adsorbents are most likely to occur on this atom. For all surfaces, the band gap that was present in the bulk almost disappeared.
 | ||
| Fig. 12 Projected DOS over the atoms of the surfaces: (a) (001); (b) (010); (c) (100) plotted using a Gaussian width of 0.005 Ry. | ||
4. Conclusion
The structural and electronic properties of the arsenopyrite and its different cleavage surfaces have been investigated by a DFT/plane waves method. For the arsenopyrite bulk, the structural parameters are in good agreement with the experimental data and previously reported calculated values. The Bader’s QTAIM analysis indicates that there is no Fe–Fe bond in arsenopyrite. Only a ring critical bond was found between the two Fe atoms. The As–S bond has covalent character and the Fe–As and Fe–S bonds have ionic character. In agreement to the previously reported calculations of covellite, pyrite and marcasite, which concluded that the S–S bond is unlikely to be broken, the As–S bond is also unlikely to be broken in the cleavage. The surface and cleavage energies have been estimated for different surfaces. The (001), (010) and (100) planes are considered the most favored cleavage planes, with surface energies close to 1.07 J m−2. In these three surfaces the As–S bond is not broken and the Fe, As and S atoms are exposed on the surface. The (001), (010) and (100) surfaces are adequate models for investigating the surface reactivity of arsenopyrite. The projected DOS on the surface show that the valence and conduction bands close to the Fermi level are mostly due to the d-orbitals of iron atoms indicating that this atom is the preferred site for adsorption and, hence, must be involved in the initial steps of the arsenopyrite oxidation. The surface oxidation of arsenopyrite is presently being investigated in our laboratory.Acknowledgements
We would like to thank Prof. Renata Diniz, Prof. Claudio de Oliveira and Dr Angel Morales-García for the fruitful discussions. The support of the Brazilian agencies Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Conselho Nacional para o Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) are also gratefully acknowledged. The National Institute of Science and Technology for Mineral Resources, Water and Biodiversity has also supported this work – INCT-ACQUA (http://www.acqua-inct.org).References
- H. W. Nesbitt, I. J. Muir and A. R. Prarr, Oxidation of arsenopyrite by air and air-saturated, distilled water, and implications for mechanism of oxidation, Geochim. Cosmochim. Acta, 1995, 59(9), 1773–1786 CrossRef CAS.
- C. L. Corkhill and D. J. Vaughan, Arsenopyrite oxidation – A review, Appl. Geochem., 2009, 24(12), 2342–2361 CrossRef CAS.
- F. Rull, et al. Spectroscopic Raman study of sulphate precipitation sequence in Rio Tinto mining district (SW Spain), Environ. Sci. Pollut. Res., 2014, 21(11), 6783–6792 CrossRef CAS PubMed.
- A. Akcil and S. Koldas, Acid Mine Drainage (AMD): causes, treatment and case studies, J. Cleaner Prod., 2006, 14(12–13), 1139–1145 CrossRef.
- H. F. Cheng, et al. Geochemical processes controlling fate and transport of arsenic in acid mine drainage (AMD) and natural systems, J. Hazard. Mater., 2009, 165(1–3), 13–26 CrossRef CAS PubMed.
- A. N. Buckley and G. W. Walker, The Surface-Composition of Arsenopyrite Exposed to Oxidizing Environments, Appl. Surf. Sci., 1988, 35(2), 227–240 CrossRef CAS.
- P. G. Fernandez, H. G. Linge and M. W. Wadsley, Oxidation of arsenopyrite (FeAsS) in acid.1. Reactivity of arsenopyrite, J. Appl. Electrochem., 1996, 26(6), 575–583 CrossRef CAS.
- H. W. Nesbitt and I. J. Muir, Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air, Mineral. Petrol., 1998, 62(1–2), 123–144 CrossRef CAS.
- C. M. V. B. Almeida and B. F. Giannetti, Electrochemical study of arsenopyrite weathering, Phys. Chem. Chem. Phys., 2003, 5(3), 604–610 RSC.
- Y. L. Mikhlin, A. S. Romanchenko and I. P. Asanov, Oxidation of arsenopyrite and deposition of gold on the oxidized surfaces: a scanning probe microscopy, tunneling spectroscopy and XPS study, Geochim. Cosmochim. Acta, 2006, 70(19), 4874–4888 CrossRef CAS.
- C. L. Corkhill, et al. The oxidative dissolution of arsenopyrite (FeAsS) and enargite (Cu3AsS4) by Leptospirillum ferrooxidans, Geochim. Cosmochim. Acta, 2008, 72(23), 5616–5633 CrossRef CAS.
- A. Hung, et al. Density-functional theory studies of pyrite FeS2(100) and (110) surfaces, Surf. Sci., 2002, 513(3), 511–524 CrossRef CAS.
- A. Hung, et al. Density-functional theory studies of pyrite FeS2 (111) and (210) surfaces, Surf. Sci., 2002, 520(1–2), 111–119 CrossRef CAS.
- A. Stirling, M. Bernasconi and M. Parrinello, Ab initio simulation of water interaction with the (100) surface of pyrite, J. Chem. Phys., 2003, 118(19), 8917–8926 CrossRef CAS.
- A. Stirling, M. Bernasconi and M. Parrinello, Defective pyrite (100) surface: An ab initio study, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(16), 165406 CrossRef.
- P. H. L. Sit, M. H. Cohen and A. Selloni, Interaction of Oxygen and Water with the (100) Surface of Pyrite: Mechanism of Sulfur Oxidation, J. Phys. Chem. Lett., 2012, 3(17), 2409–2414 CrossRef CAS PubMed.
- C. de Oliveira and H. A. Duarte, Disulphide and metal sulphide formation on the reconstructed (001) surface of chalcopyrite: a DFT study, Appl. Surf. Sci., 2010, 257(4), 1319–1324 CrossRef CAS.
- G. F. de Lima, et al. Water Adsorption on the Reconstructed (001) Chalcopyrite Surfaces, J. Phys. Chem. C, 2011, 115(21), 10709–10717 Search PubMed.
- C. de Oliveira, et al. Reconstruction of the Chalcopyrite Surfaces—A DFT Study, J. Phys. Chem. C, 2012, 116(10), 6357–6366 CAS.
- G. F. de Lima, et al. Sulfuric and hydrochloric acid adsorption on the reconstructed sulfur terminated (001) chalcopyrite surface, Int. J. Quantum Chem., 2012, 112(19), 3216–3222 CrossRef.
- C. L. Corkhill, M. C. Warren and D. J. Vaughan, Investigation of the electronic and geometric structures of the (110) surfaces of arsenopyrite (FeAsS) and enargite (Cu3AsS4), Mineral. Mag., 2011, 75(1), 45–63 CrossRef CAS.
- M. Reich and U. Becker, First-principles calculations of the thermodynamic mixing properties of arsenic incorporation into pyrite and marcasite, Chem. Geol., 2006, 225(3–4), 278–290 CrossRef CAS.
- M. Blanchard, et al. Arsenic incorporation into FeS2 pyrite and its influence on dissolution: a DFT study, Geochim. Cosmochim. Acta, 2007, 71(3), 624–630 CrossRef CAS.
- C. I. Pearce, R. A. D. Pattrick and D. J. Vaughan, Electrical and Magnetic Properties of Sulfides, Rev. Mineral. Geochem., 2006, 61(1), 127–180 CrossRef CAS.
- C. Klein, C. S. Hurlbut and J. D. Dana, Manual of mineralogy, (after James D. Dana), J. Wiley, New York, 21st edn, 1999, vol. xiv, p. 681, 4p. of col. plates Search PubMed.
- H. Fuess, et al. Crystal-Structure Refinement and Electron-Microscopy of Arsenopyrite, Zeitschrift Fur Kristallographie, 1987, vol. 179, 1–4, pp. 335–346 Search PubMed.
- N. Morimoto and L. A. Clark, Arsenopyrite Crystal-Chemical Relations, Am. Mineral., 1961, 46(11–2), 1448–1469 CAS.
- D. J. Vaughan and J. R. Craig, Mineral chemistry of metal sulfides, Cambridge earth science series, Cambridge University Press, Cambridge Eng., 1978, New York, vol. xv, p. 493 Search PubMed.
- L. Bindi, et al. Stoichiometric Arsenopyrite, FeAsS, from La Roche-Balue Quarry, Loire-Atlantique, France: Crystal Structure and Mossbauer Study, Can. Mineral., 2012, 50(2), 471–479 CrossRef CAS.
- M. Ford and C. C. Ferguson, Cleavage Strain in the Variscan Fold Belt, County Cork, Ireland, Estimated from Stretched Arsenopyrite Rosettes, J. Struct. Geol., 1985, 7(2), 217–223 CrossRef.
- G. A. Wolff and J. D. Broder, Cleavage and the Identification of Minerals, Am. Mineral., 1960, 45(11–2), 1230–1242 CAS.
- E. S. Dana and W. E. Ford, A Text-book of Mineralogy: With an Extended Treatise on Crystallography and Physical Mineralogy, Wiley, New York, 4th edn, 1966 Search PubMed.
- P. Giannozzi, et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Accurate and Simple Analytic Representation of the Electron-Gas Correlation-Energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45(23), 13244–13249 CrossRef.
- D. Vanderbilt, Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41(11), 7892–7895 CrossRef.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188–5192 CrossRef.
- N. Marzari, et al. Thermal Contraction and Disordering of the Al(110) Surface, Phys. Rev. Lett., 1999, 82(16), 3296–3299 CrossRef CAS.
- D. Beeman, Some Multistep Methods for Use in Molecular-Dynamics Calculations, J. Comput. Phys., 1976, 20(2), 130–139 CrossRef.
- M. Parrinello and A. Rahman, Polymorphic Transitions in Single-Crystals – a New Molecular-Dynamics Method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91(5), 893–928 CrossRef CAS.
- A. Otero-de-la-Roza, et al. Critic: a new program for the topological analysis of solid-state electron densities, Comput. Phys. Commun., 2009, 180(1), 157–166 CrossRef CAS.
- A. Otero-de-la-Roza, D. Abbasi-Perez and V. Luana, GIBBS2: A new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation, Comput. Phys. Commun., 2011, 182(10), 2232–2248 CrossRef CAS.
- V. K. Gudelli, et al. Phase Stability and Thermoelectric Properties of the Mineral FeS2: An Ab Initio Study, J. Phys. Chem. C, 2013, 117(41), 21120–21131 CAS.
- F. Hulliger and E. Mooser, Semiconductivity in pyrite, marcasite and arsenopyrite phases, J. Phys. Chem. Solids, 1965, 26(2), 429–433 CrossRef CAS.
- W. B. Pearson, Compounds with the marcasite structure, in Zeitschrift für Kristallographie - Crystalline Materials, 1965, p. 449 Search PubMed.
- E. H. Nickel, Structural stability of minerals with the pyrite, marcasite, arsenopyrite and lollingite structures, Can. Mineral., 1968, 9(3), 311–321 CAS.
- E. H. Nickel, The application of ligand-field concepts to an understanding of the structural stabilities and solid-solution limits of sulphides and related minerals, Chem. Geol., 1970, 5(4), 233–241 CrossRef CAS.
- G. Brostigen and A. Kjekshus, Bonding Schemes for Compounds with Pyrite, Marcasite, and Arsenopyrite Type Structures, Acta Chem. Scand., 1970, 24(8), 2993–3012 CrossRef CAS.
- J. b. Goodenou, Energy-Bands in TX2 Compounds with Pyrite, Marcasite, and Arsenopyrite Structures, J. Solid State Chem., 1972, 5, 1144–1152 Search PubMed.
- L. Pauling, Metal-Metal Bond Lengths in Complexes of Transition-Metals, Proc. Natl. Acad. Sci. U. S. A., 1976, 73(12), 4290–4293 CrossRef CAS.
- J. A. Tossell, D. J. Vaughan and J. K. Burdett, Pyrite, marcasite, and arsenopyrite type minerals: Crystal chemical and structural principles, Phys. Chem. Miner., 1981, 7(4), 177–184 CrossRef CAS.
- M. S. Schmokel, et al. Atomic properties and chemical bonding in the pyrite and marcasite polymorphs of FeS2: a combined experimental and theoretical electron density study, Chem. Sci., 2014, 5(4), 1408–1421 RSC.
- R. A. Jones and H. W. Nesbitt, XPS evidence for Fe and As oxidation states and electronic states in loellingite (FeAs2), Am. Mineral., 2002, 87(11–12), 1692–1698 CrossRef CAS.
- W. Setyawan and S. Curtarolo, High-throughput electronic band structure calculations: Challenges and tools, Comput. Mater. Sci., 2010, 49(2), 299–312 CrossRef.
- D. J. Vaughan, Sulfide Mineralogy and Geochemistry: Introduction and Overview, Rev. Mineral. Geochem., 2006, 61(1), 147 Search PubMed.
- F. Tran and P. Blaha, Accurate Band Gaps of Semiconductors and Insulators with a Semilocal Exchange-Correlation Potential, Phys. Rev. Lett., 2009, 102(22), 226401 CrossRef PubMed.
- I. J. Ferrer, et al. About the band gap nature of FeS2 as determined from optical and photoelectrochemical measurements, Solid State Commun., 1990, 74(9), 913–916 CrossRef CAS.
- A. Ennaoui, et al. Iron disulfide for solar energy conversion, Sol. Energy Mater. Sol. Cells, 1993, 29(4), 289–370 CrossRef CAS.
- I. Opahle, K. Koepernik and H. Eschrig, Full-potential band-structure calculation of iron pyrite, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(20), 14035–14041 CrossRef CAS.
- G. V. Gibbs, et al. Theoretical Electron Density Distributions for Fe- and Cu-Sulfide Earth Materials: A Connection between Bond Length, Bond Critical Point Properties, Local Energy Densities, and Bonded Interactions, J. Phys. Chem. B, 2007, 111(8), 1923–1931 CrossRef CAS PubMed.
- Y. Aray, et al. Correlation of the Topology of the Electron Density of Pyrite-Type Transition Metal Sulfides with Their Catalytic Activity in Hydrodesulfurization, Angew. Chem., Int. Ed., 2000, 39(21), 3810–3813 CrossRef CAS.
- A. Morales-García, et al. First-Principles Calculations and Electron Density Topological Analysis of Covellite (CuS), J. Phys. Chem. A, 2014, 5823–5831 Search PubMed.
- P. Mori-Sánchez, A. M. Pendás and V. Luaña, A Classification of Covalent, Ionic, and Metallic Solids Based on the Electron Density, J. Am. Chem. Soc., 2002, 124(49), 14721–14723 CrossRef.
- D. W. Fan, et al. X-ray diffraction study of arsenopyrite at high pressure, Phys. Chem. Miner., 2011, 38(2), 95–99 CrossRef CAS.
- T. Chattopadhyay and H. G. Vonschnering, High pressure X-ray diffraction study on p-FeS2, m-FeS2 and MnS2 to 340 kbar: a possible high spin-low spin transition in MnS2, J. Phys. Chem. Solids, 1985, 46(1), 113–116 CrossRef CAS.
- Y. Le Page and J. R. Rodgers, Ab initio elasticity of FeS2 pyrite from 0 to 135 GPa, Phys. Chem. Miner., 2005, 32(8–9), 564–567 CrossRef CAS.
- S. Merkel, et al. Equation of state, elasticity, and shear strength of pyrite under high pressure, Phys. Chem. Miner., 2002, 29(1), 1–9 CrossRef CAS.
- P. W. Tasker, The stability of ionic crystal surfaces, J. Phys. C: Solid State Phys., 1979, 12(22), 4977 CrossRef CAS.
- R. Dovesi, et al. Ab Initio Quantum Simulation in Solid State Chemistry, in Reviews in Computational Chemistry, John Wiley & Sons, Inc., 2005, pp. 1–125 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table with the surfaces corresponding to the C21/d and P21/c unit cells, QTAIM critical points, the K-points used for band calculations, project DOS over the arsenopyrite atoms and the cleavage surfaces are available. See DOI: 10.1039/c4ra13807d |
| This journal is © The Royal Society of Chemistry 2015 |