
Recent developments in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts: a review
Shuying Dong
a,
Jinglan Fenga,
Maohong Fan
cd,
Yunqing Pia,
Limin Hua,
Xiao Hana,
Menglin Liua,
Jingyu Sun
*b and
Jianhui Sun
*a
aSchool of Environment, Henan Normal University, Key Laboratory for Yellow River and Huai River Water Environmental and Pollution Control, Ministry of Education, Henan Key Laboratory for Environmental Pollution Control, Xinxiang, Henan 453007, P. R. China. E-mail: sunjh@htu.cn; Tel: +86-373-3325971
bCenter for Nanochemistry (CNC), College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: sunjy-cnc@pku.edu.cn; Tel: +86-10-62757157
cDepartment of Chemical and Petroleum Engineering, University of Wyoming, 1000 E University Avenue, Laramie, WY 82071, USA
dSchool of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
First published on 7th January 2015
Abstract
Visible light-responsive photocatalytic technology holds great potential in water treatment to enhance purification efficiency, as well as to augment water supply through the safe usage of unconventional water sources. This review summarizes the recent progress in the design and fabrication of visible light-responsive photocatalysts via various synthetic strategies, including the modification of traditional photocatalysts by doping, dye sensitization, or by forming a heterostructure, coupled with π-conjugated architecture, as well as the great efforts made within the exploration of novel visible light-responsive photocatalysts. Background information on the fundamentals of heterogeneous photocatalysis, the pathways of visible light-responsive photocatalysis, and the unique features of visible light-responsive photocatalysts are presented. The photocatalytic properties of the resulting visible light-responsive photocatalysts are also covered in relation to the water treatment, i.e., regarding the photocatalytic degradation of organic compounds and inorganic pollutants, as well as photocatalytic disinfection. Finally, this review concludes with a summary and perspectives on the current challenges faced and new directions in this emerging area of research.
 Shuying Dong | Dr Shuying Dong received her PhD in Environmental Sciences in 2014 at the School of Environment, Henan Normal University, China, under the supervision of Dr Jianhui Sun. She has engaged in research in the field of photocatalysis for water treatment for more than seven years and has published 13 research papers in peer-reviewed journals in such an area. She is currently an assistant professor at the School of Environment, Henan Normal University. Her current research interests center on the rational synthesis and mechanism exploration of visible light-responsive photocatalysts for wastewater treatment. |
 Maohong Fan | Dr Maohong Fan is an SER Professor in Chemical and Petroleum Engineering at the University of Wyoming, USA. He has led and worked on many projects in the areas of chemical production, clean energy generation, and environmental protection, which have been supported by various domestic and international funding agencies, such as NSF, DOE and EPA in the USA, and industrial companies, such as Siemens and Caterpillar. He has published over 175 refereed books, book chapters, and papers in different chemical and environmental engineering, energy, and chemistry journals. |
 Jingyu Sun | Dr Jingyu Sun received a BSc in Materials Sciences and Engineering from Zhejiang University, China in 2008, where he obtained the Honors degree from the Mixed Class, Chu Kochen Honors College. He obtained his DPhil in 2013 from the Department of Materials, University of Oxford, UK, for his work on the controlled growth of carbon nanomaterials on perovskite substrates. He is currently a postdoctoral associate in the Center for Nanochemistry, Peking University, where he works on graphene synthesis and related applications. |
 Jianhui Sun | Dr Jianhui Sun (Professor, Henan Normal University, China) obtained his PhD in Environmental Sciences at the Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, China. His water environment and pollution control research laboratory at the School of Environment, Henan Normal University is active in the field of photocatalysis, advanced oxidation technology, wastewater treatments and developing new approaches/concepts suitable for water and energy related applications. He has published over 50 research papers in peer-reviewed journals of international repute (h-index = 18). Dr Sun has teaching experience in Environmental Monitoring and Environmental Pollution Control. |
1. Introduction
With the rapid development of industrialization and urbanization, as well as huge population increases, the urgent demand for, but acute shortage of, clean water sources have attracted immense attention all over the world. This difference in clean water demand and availability is expected to continue to deteriorate with respect to growing contamination, owing to the overwhelming discharge of contaminants and pollutants into the natural water cycle. As far as the environment is concerned, the reuse and recycling of wastewater effluents is thus necessary to augment the limited fresh water supply and to offset more possible water resources in the long run.1 During the past few decades, a variety of practical strategies have been implemented to develop viable wastewater treatment technologies.2–6 For instance, conventionally, biological treatments were designed to effectively eliminate assorted types of contaminants from wastewater in the short term; however, these techniques also normally lead to the production of secondary pollution,7 some of which even involves the presence of health-threatening bacteria and soluble refractory organic compounds that are hard to remove.8 In this regard, developing nondestructive, green, and sustainable methodologies for water treatment is hence of great significance.To date, photocatalysis has been considered as one of the most appealing options for wastewater treatment, due to its great potential and high efficiency by using sunlight to remove organic pollutants and harmful bacteria with the aid of a solid photocatalyst.9,10 Here, it is well known that the photocatalytic efficiency is not only influenced by the nature of the employed photocatalysts, but is also affected by the irradiated light source. Generally, the catalyst can be photo-activated by a photon with energy equal to or higher than its band-gap energy (Eg). A stronger irradiation intensity would normally induce more efficient photolysis reactions.11 As a safe and renewable energy source, natural sunlight is the ideal source to supply energy for these activation processes. Moreover, solar energy possesses special advantages, such as its cleanness and abundance. The sun delivers about four-orders of magnitude larger energy to the earth's surface per year than the energy annually used by humans all over the world.12,13 To better utilize this solar energy to tackle water contamination issues, it is strongly desirable to explore novel catalysts with visible light-driven photocatalytic performance.
TiO2 is the most widely studied photocatalyst, due to its cost-effectiveness, nontoxicity, and unique photocatalytic efficiency, as well as high stability. The basic mechanism of the photocatalytic degradation of pollutants using TiO2 deals involves the absorption of near-UV light by TiO2, the input of the light energy to induce charge separation to form electron (e−)–hole (h+) pairs, and the participation of e− and h+ in the oxidation–reduction reactions with suitable substrates.14 However, there are a number of drawbacks of pure TiO2 in photocatalysis, including the relatively large band-gap and the related fact that it can only absorb a small portion of UV radiation.15 To facilitate the usage of energy from natural sunlight, certain routes have been attempted to modify TiO2 accordingly, including non-/transition-metal ion doping,16,17 sensitization,18 ion implantation,19 narrower band-gap semiconductor coupling,20 and π-conjugated structure compositing,21 etc. For example, Rey et al.16 synthesized TiO2–WO3 photocatalysts, which exhibited higher catalytic activity than that of pure TiO2 for the removal of a mixture of emerging contaminants through photocatalytic ozonation under visible light radiation. A study conducted by our group demonstrated that nitrogen-doped TiO2 showed superior photocatalytic performance than that of the commercial Dugussa P25 TiO2 for the degradation of a specific azo dye (Orange G) under visible light irradiation, which was attributed to the fact that nitrogen doping gave rise to the appearance of a new absorption band in the visible region.17
There has been a growing interest lately in the development of directly visible light-responsive photocatalysts for water treatment, partially due to the complex and costly steps involved in the modification of the UV-illuminated ones.22 To this end, a plethora of photocatalysts have been created, e.g., SnS2,23 CuO/BiVO4,24 ZnS,25 g-C3N4/BiPO4,26 AgBr/Ag3PO4/MWCNTs,27 Ag3PO4,28 BiOX (X = Cl, Br, I),29 Bi2MO6 (M = W, Mo),30,31 and MWO4 (M2+ = Co, Cu, Pb, Cd, Mn and Zn),32 ZnSnO3,33 MVO4 (M = Bi, Sm),34,35 nanostructured iron(III) oxides,36 and oxynitride,37 where considerable advances with regard to the photocatalytic performances have been steadily achieved, opening up the possibility for the practical usage of natural solar energy for wastewater degradation, as well as for environmental protection. For example, Mondal et al.23 demonstrated that the dosage of variable concentrations of thioacetamide enabled the congregation of SnS2 nanoflakes to nanoflowers and nanoyarns, which were proven to be promising for promoting the photoreduction of toxic Cr(VI) wastewater under visible light irradiation. Moreover, the performance of the SnS2 nanoflower was found to be superior to that of the nanoyarn, owing to the increased surface area and higher pore volume. Sheng et al.30 probed the photocatalytic activities of Bi2WO6 under visible light conditions using phenol degradation as a model reaction, where the observations obtained by employing spin-trapping electron paramagnetic resonance spectroscopy indicated that the irradiated Bi2WO6 was responsible not only for the production of ˙OH radicals (via water oxidation), but also for the generation of H2O2 (via a two-electron reduction of O2).
From a practical application point of view, solid-substrate-supported photocatalysts have in particular been recognized as an important class of industrial catalysts that are closely related with versatile key technologies in water treatment. These supported catalysts were found to possess several advantages, such as improved resistance to agglomeration, good accessibility of the substrate molecules, site isolation, practical usage in continuous-flow systems, and mechanical robustness, etc.19,22,38–41 Moreover, such materials enabled the direct integration of technological devices to improve the photocatalysts' activity.39 For example, Carraro et al.40 prepared polymorphous iron(III) oxide nanosystems [α-Fe2O3 (hematite) and β-Fe2O3 (bixbyite)] via a chemical vapor deposition approach, which were well characterized and tested in the photo-degradation of methylene blue (MB) in the liquid phase under simulated solar light irradiation. The obtained results showed a significant dependence of the purity and morphology on the synthetic conditions, and where the surface area of the sample appeared to have a critical influence on the photocatalytic performance. Aziz et al.41 fabricated a magnetically separable TiO2 nanocomposite with SiO2 coating (supported on a permanent magnet NiFe2O4), where the prepared TiO2 material featured a lower band-gap energy (2.26 eV) and higher visible light absorption than that of pure TiO2 (2.76 eV). Accordingly, the nanocomposite exhibited improved sunlight activity for the photodegradation of 2,4-dichlorophenol, as well as a good stability against the loss of its magnetic property for reuse.
This field of research has stimulated great efforts on the preparation, modification, and application of visible light-responsive photocatalysts, resulting in many important findings being reported during the past few years. Many insightful review articles have dealt with the target synthesis, theoretical investigation, photocatalysis design, and possible applications of photocatalysts, especially focusing on the specific modification of certain types of photocatalysts. In contrast, few reviews are concerned with the discussion of photocatalysis in the broader context of visible light-responsive photocatalyst types, i.e., the routes to affording the visible light responses, the effects of the corresponding properties, and their related application within water treatment. Based on the aforementioned facts, the present review summarizes the latest developments in visible light-responsive photocatalysis, with a focus on the broad coverage of the employed catalyst systems. The review starts with a brief introduction to the fundamentals and speculated mechanisms of visible light-responsive photocatalysis, followed by a discussion of the recent achievements made in the design, modification, and applications of visible light-responsive photocatalyst systems. Finally, the future outlooks and perspectives are also considered.
2. Fundamentals and proposed pathways of visible light-responsive photocatalysis
In general, the system of heterogeneous photocatalysis using semiconductor materials consists of a light harvesting antenna and several active species to facilitate the pollutant degradation. The series of chain oxidative–reductive reactions that occur at the photon-activated surface has been broadly proposed as:| Photocatalyst + hν → h+ + e− | (2.1) |
| h+ + H2O → ˙OH + H+ | (2.2) |
| h+ + OH− → ˙OH | (2.3) |
| h+ + pollutant → (pollutant)+ | (2.4) |
| e− + O2 → ˙O2− | (2.5) |
| ˙O2− + H+ → ˙OOH | (2.6) |
| 2˙OOH → O2 + H2O2 | (2.7) |
| H2O2 + ˙O2− → ˙OH + OH− + O2 | (2.8) |
| H2O2 + hν → 2˙OH | (2.9) |
| Pollutant + (˙OH, h+, ˙OOH or O2−) → degradation product | (2.10) |
When the semiconductor is irradiated by an input light possessing an ultra-band-gap energy (hν > Eg), a valence band (VB) electron (e−) is excited to the conduction band (CB), leaving behind a photogenerated hole (h+) at the VB. Accordingly, the produced e−/h+ pairs are able to migrate to the surface of the semiconductor and participate in redox reactions. The photocatalytic reaction usually involves three main active species: a hydroxyl radical (˙OH), h+, and a superoxide radical (˙O2−), where ˙OH is the primary oxidant in the photocatalytic degradation of the pollutant in the aqueous solution. The generation of ˙OH radicals normally via two routes, (i) H2O and OH− in the water environment are readily oxidized by photogenerated h+ to form ˙OH radicals; (ii) O2 presented in the aqueous solution is reduced by photogenerated e− to form ˙O2− radicals, followed by reacting with h+ (forming ˙OOH radicals) and then further decomposition to produce ˙OH radicals. Moreover, the photogenerated h+ is widely considered as an oxidant for directly degrading organic contaminates, the capacity of which depends on the catalyst type and oxidation conditions.42 It is to be noted that the photo-induced e− can easily recombine with h+ after their generation in the absence of electron or hole scavengers. In this regard, the presence of specific scavengers is vital for suppressing the charge recombination rates and for enhancing the efficiency of photocatalysis.
To design a photocatalyst capable of utilizing safe and sustainable solar energy effectively, several critical requirements need to be satisfied. First, the semiconductor material should have a smaller band-gap to allow it to absorb solar energy across a broad range of spectrum. Simultaneously, the semiconductor should have a relatively positive enough valence band for the ample production of h+ and ˙OH radicals.43 Second, the catalyst should possess a particular platform/system for the efficient charge separation and transportation.44,45 Moreover, the semiconductor materials should have good photoelectrochemical stability in the electrochemical reactions.46
Generally, along with the electronic band structures, other features such as the material choice, morphological architecture, crystallinity, and surface properties should also be taken into consideration when building up an efficient and stable visible light-responsive photocatalytic system.47,48 The choice of the semiconductor materials is particularly important, since it determines the level of the visible light response and, hence, the overall efficiency. The right morphological architecture with a short distance between the photocarrier-generating junction and the redox reaction center can effectively improve the carrier separation and transportation.49 Moreover, a high degree of crystallinity with crystal defects would minimize the interface recombination, thereby enhancing the efficiencies of the photogenerated electrons and holes to participate in the desired reactions.50 The surface area of the photocatalysts, which depends upon the porosity and geometrical shape of the materials, also exerts a crucial effect on the photocatalytic activity, owing to the fact that the adsorption of pollutants is a critical step.51
3. Overview of visible light-responsive photocatalysts
3.1. Modification routes towards augmenting the visible light response of photocatalysts
Compared to other photocatalysts for wastewater treatment, TiO2 and ZnO have received considerable attention, due to their advanced photocatalytic performance under UV light irradiation.52,53 The drawback of these semiconductors, however, lies in the fact that they are inactive under visible light irradiation, due to their large band-gap, which impedes their usage as solar energy harvesting photocatalysts. To overcome such a vital deficiency, certain modifications toward the semiconductors system have been attempted to enable their visible light responses with good efficiency.Nitrogen doping has been the most intensively studied amongst all the non-metal dopants. Wang et al.58 prepared N-doped TiO2 by the heat treatment of commercial P25-TiO2 in a NH3 gas flow, and the product was characterized by a series of techniques to investigate the origin of the visible light response of N-doped TiO2. The results indicated that N-doped TiO2 possessed triplet g value electron spin resonance signals (g = 1.987, 2.004, and 2.024) and visible light absorption in a wavelength range of 400–520 nm, which were attributed to the formation of single-electron-trapped oxygen vacancies (denoted as SETOVs) in a certain chemical environment. The visible light photocatalytic activity of N-doped TiO2 was co-determined by the formation of SETOVs in the TiO2 matrix and by the existence of doped-N on the surface. Similarly, one of our previous studies also reported that N-doped TiO2 showed higher photocatalytic activity than that of commercial Dugussa P25 TiO2 under visible light irradiation, which could be due to the presence of a TiO2 anatase structure, and a new absorption band in the visible region caused by the nitrogen doping.17
Carbon doping has been explored with consideration of its low cost and its potential for band-gap narrowing, which can help to achieve significant improvements in visible light absorption capabilities.59–62 Chen et al.60 synthesized pure anatase C-doped TiO2 by a low-pressure flat-flame metal organic chemical vapor condensation method by eliminating the nitrogen doping possibility. They demonstrated that visible light absorption was attributed to the carbon doping, but that the carbon did not incorporate into the TiO2 crystal, and instead located on the surface. They also claimed that the C–C bond was responsible for the light absorption. Dai and colleagues61 prepared porous C-doped Bi2O3 with a high visible light-responsive photocatalytic activity via a simple calcination of bismuth nitrate pentahydrate in a glycol solution. It was shown that the carbon was incorporated into the lattice of Bi2O3, as the absorption edge of C-doped Bi2O3 had an obvious red shift with augmented absorption intensity in the region of 450–530 nm, which was responsible for the enhanced photocatalytic activity over the pure one. Samadi and coauthors62 utilized an electrospinning technique to fabricate multi-walled carbon nanotubes (MWCNTs)-doped ZnO nanofibers with visible light-responsive photocatalytic activity. Their study revealed that Zn–O–C bonds were formed and the energy band-gap of the composite was 2.94 eV, which was lower than that of the pure ZnO nanofiber (3.11 eV). Accordingly, a 7-fold enhancement in the photocatalytic activity was observed because of the delayed electron–hole recombination exerted by the synergistic effect between the MWCNTs and ZnO. However, compared to the N-doped photocatalysts, C-doped ones are considered to be more difficult to synthesize and hence have not been widely employed to date.63
The modifications of photocatalyst with noble and other metals such as Pt, Au, Ag, Cu, V, Ni, and Sn have also enabled the extension of the spectral response of photocatalysts well into the visible region.64 However, a few studies have claimed that several metallic species (especially transition metals) may act as recombination sites for the photo-induced charge carriers, thereby lowering the quantum efficiency.65 For instance, Li et al.65 fabricated hierarchical V-doped rutile TiO2 nanofibers by a flame burning method, which showed a higher doping level of V into the TiO2 crystal lattices than that prepared by calcination treatments. However, the photocatalytic activity of the synthesized V-doped rutile TiO2 nanofibrous have not yet been obviously enhanced due to the V doping, but this could be attributed to the fact that the V dopants served as electron–hole recombination centers. Vijayan et al.66 prepared visible light-activated Pt–TiO2 nanotubes by a hydrothermal technique, where Pt doping affected the morphology of the TiO2 nanotubes. Their study revealed that Pt nanoparticles were uniformly distributed on the nanotube surface and that the doping by Pt enhanced the visible light photoactivity of TiO2 nanotubes for the photo-oxidation of acetaldehyde. Electron paramagnetic resonance spectra revealed that coordinated sites and oxygen deficiency were created on the surface of the TiO2 nanotube after calcination in a hydrogen atmosphere, which further interacted with the Pt centers to alter the electronic, optical, and chemical behaviors of the TiO2 nanotube. One of our recent studies reported the synthesis of Sn-doped ZnO photocatalysts with augmented sunlight photocatalytic activity through a microwave-assisted route. The microstructure, morphology, and optical properties of the ZnO were greatly changed by the Sn doping, contributing to an enhanced sunlight photocatalytic activity (e.g., 13% higher decolorization rate and 29–52% greater mineralization efficiency than that of pure ZnO for the degradation of MB solution).67
Co-doping with two or more suitable heteroatoms (non-metal–non-metal, metal–non-metal, and metal–metal) can lead to substantial synergistic effects with respect to changing the band structures (including the CB and VB levels) of the systems to obtain the desired photocatalytic redox ability and selectivity, to enhance the visible light harvesting and charge mobility, or to modify the morphological characteristics. Moafi and colleagues68 prepared La–Zr-doped ZnO nanocomposites using a modified sol–gel method. Their characterization indicated that La–Zr-doped ZnO exhibited a smaller particle size than that of pure ZnO, and had a red-shift feature in the absorption band. Interestingly, the co-doping with La and Zr gave rise to the band-gap narrowing, as well as to an enhancement of the photo-activity. Wu et al.69 synthesized C–N co-doped TiO2 hierarchical spheres via a direct chelating process with the aid of several types of amine agents, where it was found that the optimal C and N doping concentrations were produced by using trimethylamine, which effectively reduced the band-gap of TiO2 to 2.85 eV without affecting its crystallization. Moreover, it exhibited an eightfold photocatalytic activity higher than that of commercial Degussa P25 powders for the decomposition of rhodamine B (RhB). Sun et al.70 prepared N–TiO2, Pt–TiO2, N–Fe–TiO2, N–Ni–TiO2, N–Ag–TiO2, and N–Pt–TiO2 photocatalysts by acid-catalyzed sol–gel processes, and further evaluated the corresponding photocatalytic activities via the photodegradation of phenol solutions under simulated sunlight irradiation. It is worth noting that certain types of transition metals (Fe and Ni in this case) exerted a negative effect on N-metal co-doped TiO2 photocatalysis, whilst noble metals (Ag and Pt) showed an augmentation in photocatalysis. In particular, N–Pt–TiO2 exhibited a six times higher photocatalytic efficiency than that of Degussa P25 under simulated sunlight irradiation. The synergistic effect of N–Pt co-doping was ascribed to the multivalent states of Pt.
| Dye (D) + hν → dye* (D*) | (3.1) |
| Photocatalyst + D* → ˙D+ + photocatalyst (e−) | (3.2) |
| Photocatalyst (e−) + O2 → photocatalyst + ˙O2− | (3.3) |
| e− + ˙D+ → D | (3.4) |
| ˙O2− + H+ → ˙OOH | (3.5) |
| 2˙O2− + 2H+ → ˙O2 + H2O2 | (3.6) |
| H2O2 + ˙O2− → ˙OH + OH− + O2 | (3.7) |
| Pollutant + (˙OH, ˙OOH or O2−) → degradation product | (3.8) |
The dye can absorb visible light to reach an excited state, which, in general, has a lower redox potential than that of the corresponding ground state. When the redox potential is lower than the CB of the semiconductor, the cationic radicals and conduction band electron can be easily formed if an electron is injected into the conduction band from the excited state.71,72 Li et al.72 showed that the photo-responses of the squarylium dye (ISQ) sensitized TiO2 nanoparticles were remarkably extended to the visible light region, and that their photocatalytic activity under visible light irradiation was significantly enhanced. In such a system, the ISQ dye on the sensitized surface of TiO2 could be easily excited from the ground state (D) to the excited state (D*) with the aid of visible light. This excited state dye species could then be converted to a semi-oxidized radical cation (˙D+) by the injection of an electron into the CB of TiO2, as the lowest unoccupied molecular orbital (LUMO) level of the ISQ dye matched well with the CB of TiO2, thus benefiting the charge transfer. Radicals such as ˙O2−, ˙OH, and ˙OOH were then produced via a series of protonation and reduction steps. Finally, the ˙OH radicals reacted with the MB molecules to produce the degradation product.
Furthermore, the transportation of molecules adsorbed on the surfaces was reduced at a dimensional level, which is beneficial to the reaction kinetics and results in the extension of the excitation energy range of a semiconductor into the visible region. This may also possess the capability to drive other reactions, and hence the sensitivity of the photocatalytic process for the removal of colored pollutants can be increased in the presence of low concentrations of colored pollutants.73 This approach is useful for treating textile wastewater, and one of the consequences of this is the direct oxidation of the dye. Shang et al.74 demonstrated that the enhanced photocatalytic activity of dye-sensitized TiO2 could be attributed to the wider absorption spectrum range and the electron transferred from the excited state of the dye molecule directly to the CB of TiO2, which results in a greater number of electrons in the CB of TiO2 and the consequent production of more active oxygen species. Li et al.75 found that sensitized ZnO microrods with porphyrin hetero-aggregates have an enhanced visible light photocatalytic activity compared to those of porphyrin-monomers–modified ZnO and pure ZnO, which could be attributed to the redox potential of the porphyrin hetero-aggregate matching well with the energy level of ZnO and the consequent promotion of the electron injection from the excited state of porphyrin into the CB of ZnO, correspondingly suppressing the electron–hole recombination rates. Yang et al.76 probed the usage of Alizarin Red S dye-sensitized nanoscale ZnO for the photocatalytic removal of Cr6+ from an aqueous solution under visible light irradiation. The apparent band-gap energy of the dye-sensitized nanoscale ZnO (2.79 eV) was narrower compared with that of nanoscale ZnO without dye sensitization (3.37 eV). It was suggested that the dye molecule, acting as an organic semiconductor, might exert an effect on the charge transition into the CB of ZnO in the excited state.
In addition to the extension of the light-response range and promotion of the separation of photon-generated carriers induced by the heterostructure nature, the photocatalytic performance of such a coupling system is crucially related to the size, shape, and surface area of the heterostructure. Wei et al.81 dispersed CdS nanoparticles within the entire surface of the TiO2 nanofibers, forming a CdS/TiO2 hierarchical heterostructure. The enhanced photocatalytic activities of the CdS/TiO2 heterostructure might have arisen from the increased surface area, extended light absorption region, and/or the favorable electron-transfer properties. Recently, Xu et al.31 teamed up the wide-band-gap Bi2O2CO3 with Bi2MoO6 to form hierarchical Bi2O2CO3/Bi2MoO6 heterostructured photocatalysts with superior visible light photocatalytic activity toward the degradation of RhB. The Bi2O2CO3/Bi2MoO6 heterostructure also displayed visible light photocatalytic activity for the destruction of E. coli, with excellent stability and recycling performance. In this regard, the photocatalytic degradation efficiency was related to the Bi/Mo molar ratio, where the highest degradation efficiency was observed with a Bi/Mo molar ratio of 2.88/1, which was approx. 55% and 97% higher than that of the pure Bi2O2CO3 microspheres and Bi2MoO6 nanoplatelets, respectively. Moreover, Zhou et al.82 claimed that energy-band matching was responsible for the enhanced photocatalytic activity within the heterostructured systems from the results obtained by carrying out a comparison study between PdO/TiO2 and Pd/TiO2 heterostructured nanobelts.
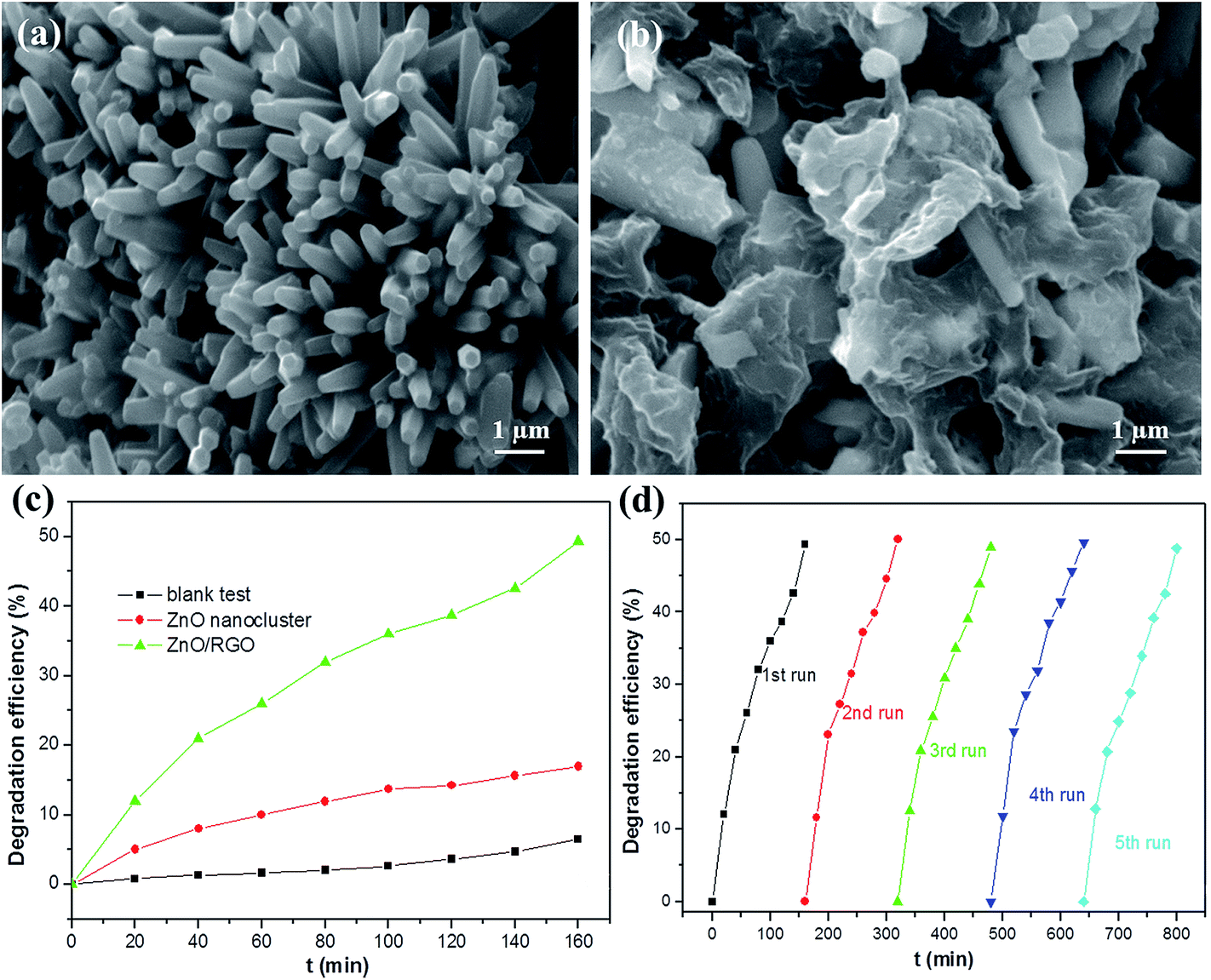 | ||
| Fig. 1 SEM images of the as-synthesized ZnO nanoclusters (a) and ZnO/RGO nanocomposites (b). (c) The degradation efficiencies of metronidazole-contained wastewater using the ZnO nanoclusters and ZnO/RGO nanocomposites under visible light irradiation. (d) Cycling runs of the ZnO/RGO nanocomposites for the photo-degradation of metronidazole. Reproduced with permission from ref. 94. | ||
C60 possessing a unique configuration and properties has been widely studied. C60 has a closed-shell configuration consisting of 30 bonding molecular orbitals with 60 π-electrons, which could efficiently cause a rapid photogenerated charge separation and a relatively slow charge recombination.95 Fu et al.86 found that C60-hybridized ZnO showed the same absorbance edge as pure ZnO, but an extended absorbance to the visible region. The photocorrosion inhibition of ZnO by coupling with C60 could be attributed to the reduced activation of the surface oxygen atom. Due to the interaction of C60 and ZnO with the aid of a conjugative π-system, a higher migration efficiency of photogenerated electrons could be produced at the interface of C60 and ZnO, which can bring about a greater photocatalytic activity for C60-hybridized ZnO. Long et al.87 reported that the photocatalytic activity of C60-incorporated TiO2 nanorods under visible light irradiation was dramatically increased by a factor of approximately 3.3 and 2.7 when compared with that of pure TiO2 nanorods and Degussa P25, respectively, which was ascribed to the effective separation of the photogenerated carriers with the introduction of C60.
As one of the π-conjugated structure materials, graphite-like carbon nitride (g-C3N4) has recently been investigated to see if it exhibited good photocatalytic activity for wastewater treatment under visible light irradiation.96,97 In comparison with the aforementioned π-conjugated structure, g-C3N4 is a soft polymer, so it should be easy to decorate on the surface of a photocatalyst, promoting the formation of core–shell structures. Sun et al.96 synthesized g-C3N4–ZnO composite photocatalysts with different ZnO dosages in wt% by a simple calcination process, the absorption edge of which shifted toward the lower energy region and longer wavelengths in comparison with that of pure ZnO and g-C3N4. The remarkable photocatalytic activity of the g-C3N4–ZnO composite for the photodegradation of methyl orange and p-nitrophenol under visible light irradiation could be mainly ascribed to the enhancement of the electron–hole separations at the interface of ZnO and g-C3N4. Fu et al.97 prepared g-C3N4–TiO2 composite samples with different weight ratios by heating mixtures of melamine and commercial TiO2. The samples with weight ratios of g-C3N4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiO2 = 2.5 exhibited the highest adsorption capacity and enhanced visible light catalytic activity for the degradation of methylene blue. The excited electrons on the surface of g-C3N4 could transfer easily to the TiO2 CB via the well-built heterojunction, correspondingly inhibiting recombination of the photogenerated electron–hole pairs.
:TiO2 = 2.5 exhibited the highest adsorption capacity and enhanced visible light catalytic activity for the degradation of methylene blue. The excited electrons on the surface of g-C3N4 could transfer easily to the TiO2 CB via the well-built heterojunction, correspondingly inhibiting recombination of the photogenerated electron–hole pairs.
Conjugated polymer modification is one of the most promising methods for modifying TiO2 to prepare visible light-responsive photocatalysts. In the combined system of a conjugated polymer and a semiconductor, it is thermodynamically possible to transfer the electrons from the conjugated polymer to the TiO2 CB under visible light irradiation, due to the fact that the conjugated polymer LUMO is energetically higher than the TiO2 CB edge. Therefore, the occurrence of interfacial charge transfer and separation between the conjugated polymer and the semiconductor guarantees an advanced photoresponse to visible light.89 Li et al.90 prepared a PANI-modified TiO2 material using an in situ chemical oxidative polymerization method, where the enhanced visible light photocatalytic activity, in terms of the phenol degradation, was due to the synergetic effect between PANI and TiO2. Meanwhile, Deng et al.91 and Luo et al.98 observed a similar trend with regard to the enhancement of photocatalytic performances compared to the bare TiO2 photocatalyst when probing the PPy-PANI-TiO2− and polyisoprene-modified TiO2 systems, respectively.
3.2. Novel visible light-responsive photocatalysts
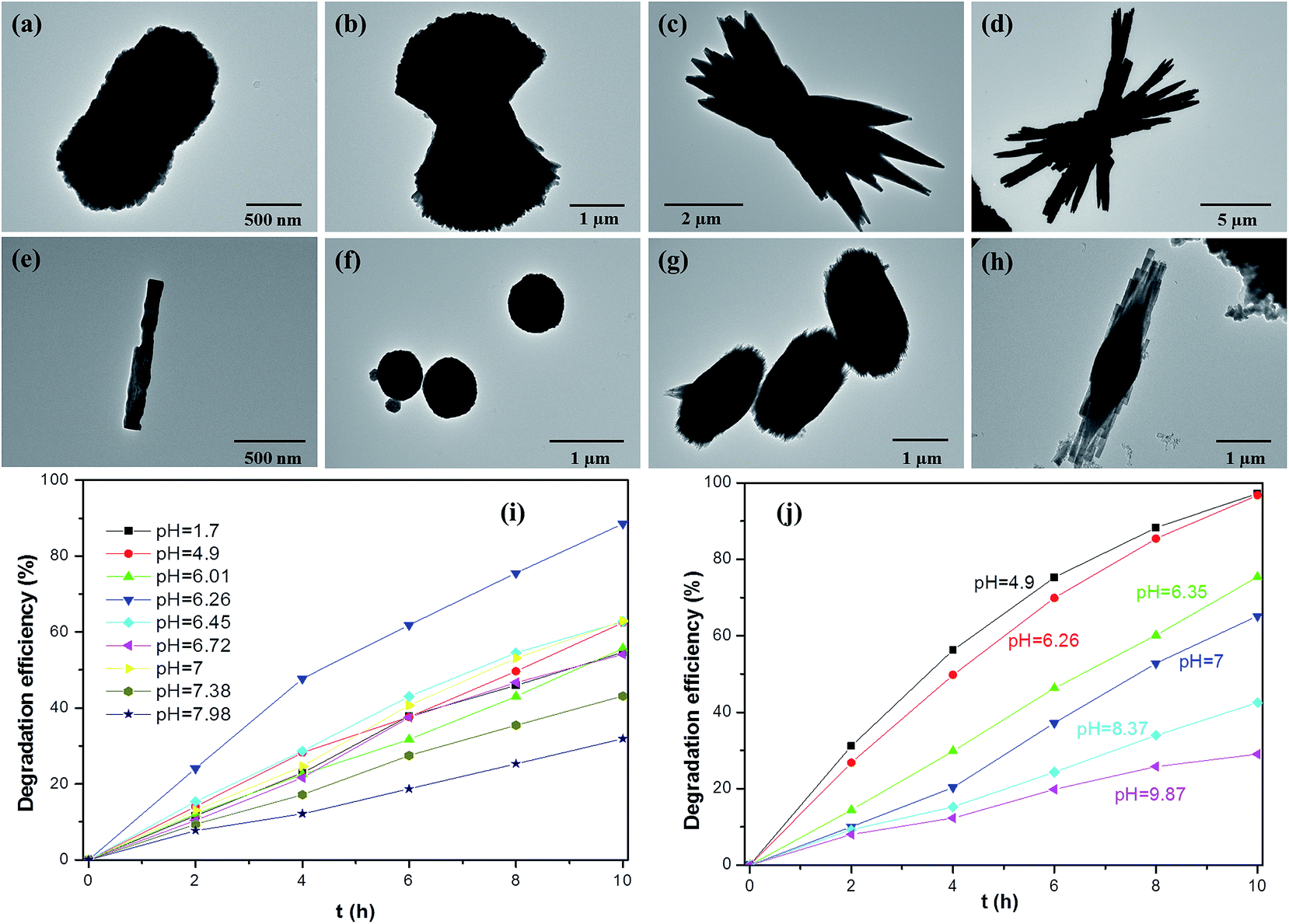 | ||
| Fig. 2 Representative TEM micrographs of different-shaped BiVO4 hierarchical structures obtained by varying the pH values of the precursors using NH3·H2O (a) pH = 4.9; (b) pH = 6.26; (c) pH = 6.72; (d) pH = 7; (e) pH = 7.3; and NaOH (f) pH = 4.9; (g) pH = 6.26; (h) pH = 7 as the pH controlling agent. The photocatalytic degradation of RhB under natural sunlight irradiation over (i) A-BiVO4 and (j) S-BiVO4 samples prepared at different pH values of the precursors. Reproduced with permission from ref. 107. | ||
The photocatalytic properties of other multi-component oxides towards the degradation of organic contaminants under visible light irradiation have been investigated. In one study, Ag3PO4 nanoparticles were synthesized by an ion-exchange reaction and nearly 80% of Cr(VI) ions were removed after visible light irradiation in Ag3PO4 suspension, where the excellent photocatalytic reduction performance was attributed to the high separation efficiency of the photogenerated charges.28 Another study was concerned with the synthesis of CaIn2O4 rods, as well as measurements of MB degradation and toluene oxidation under visible light irradiation.113 In particular, MIn2O4 (M = Ca, Sr, and Ba) semiconductors were employed for the degradation of MB under visible light irradiation, where the highest photocatalytic activity was obtained using CaIn2O4.114
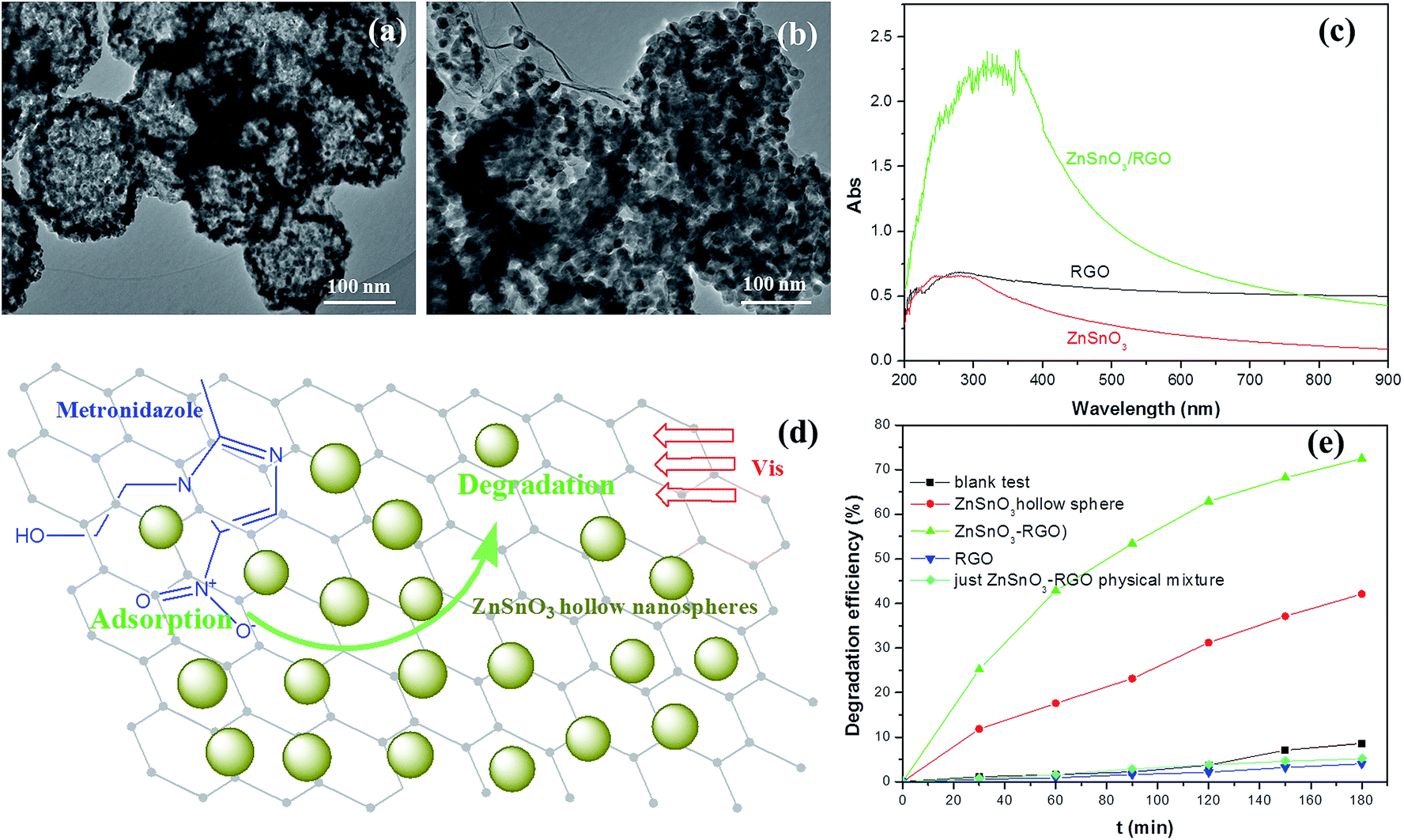 | ||
| Fig. 3 The HRTEM images of the prepared ZnSnO3 hollow nanosphere (a) and ZnSnO3/RGO nanocomposite (b). (c) The UV-Vis absorption spectra of the RGO, pure ZnSnO3 hollow nanosphere and ZnSnO3/RGO nanocomposite. The speculated illustration (d) and degradation efficiencies (e) of the metronidazole wastewater by ZnSnO3 hollow nanosphere and ZnSnO3/RGO nanocomposite photocatalysts under visible light irradiation. Reproduced with permission from ref. 33. | ||
The novel g-C3N4/SmVO4 composite photocatalyst with improved visible light photocatalytic activities for RhB degradation was investigated by Li and colleagues.35 The improvement originated from the synergetic effect of g-C3N4 and SmVO4 based on the band position; the schematic illustration of the electron–hole separation and transport at the visible light-driven g-C3N4/SmVO4 composite photocatalyst is shown in Fig. 4. Channei et al.117 showed that Fe3O4/SiO2/CeO2 core–shell magnetic structures had a higher photocatalytic degradation rate for formic and oxalic acid than that of bare CeO2 under visible light. Zhang et al.118 reported the formation of Bi2O3/Bi2SiO5 nanoheterostructures within mesoporous SiO2 microspheres, which exhibited excellent photocatalytic activities for the degradation of both acetaldehyde and bisphenol A under simulated solar light irradiation. The high photocatalytic efficiency was due to the efficiency charges separation, stemming from the heterostructure junction effect. The C60-modified Bi2MoO6 photocatalyst showed high photocatalytic activity in the reduction of bromate ions under visible light irradiation.119 The enhanced photocatalytic activity may be closely attributed to the interaction between Bi2MoO6 and C60, which increases the photo-generated electron mobility in Bi2MoO6, and as such, C60 could effectively transfer the photoelectrons from the CB of Bi2MoO6 after being illuminated under visible light irradiation. Kant et al.120 synthesized a Fe0.01Ni0.01Zn0.98O/PANI composite by an in situ free radical polymerization method. Optical and photocatalytic studies revealed that the formation of the composite further enhanced the visible light absorption and photodegradation efficiency against MB under visible light irradiation. MWCNTs have the potential for use in the fabrication of novel nanocomposites, due to their special aperture structure, high aspect ratio, and large electron storage capacity. ZnFe2O4/MWCNTs composite has been found to be a suitable visible light-responsive catalyst for the degradation of RhB.121 High-quality CuSe/ZnSe flower-like nanocomposites were fabricated as a visible light-responsive photocatalyst through utilizing the p–n junction effect and band-gap engineering. The 8 wt% CuSe/ZnSe sample exhibited over 4 times and 2.5 times degradation ratios for both MO and MB over that of the pure ZnSe sample, respectively. The improvements were attributed to the effective separation of the photogenerated carriers by the direct initiated interfacial charge transfer from the VB of ZnSe to CuSe, resulting in the reduction of CuSe to Cu2Se.122
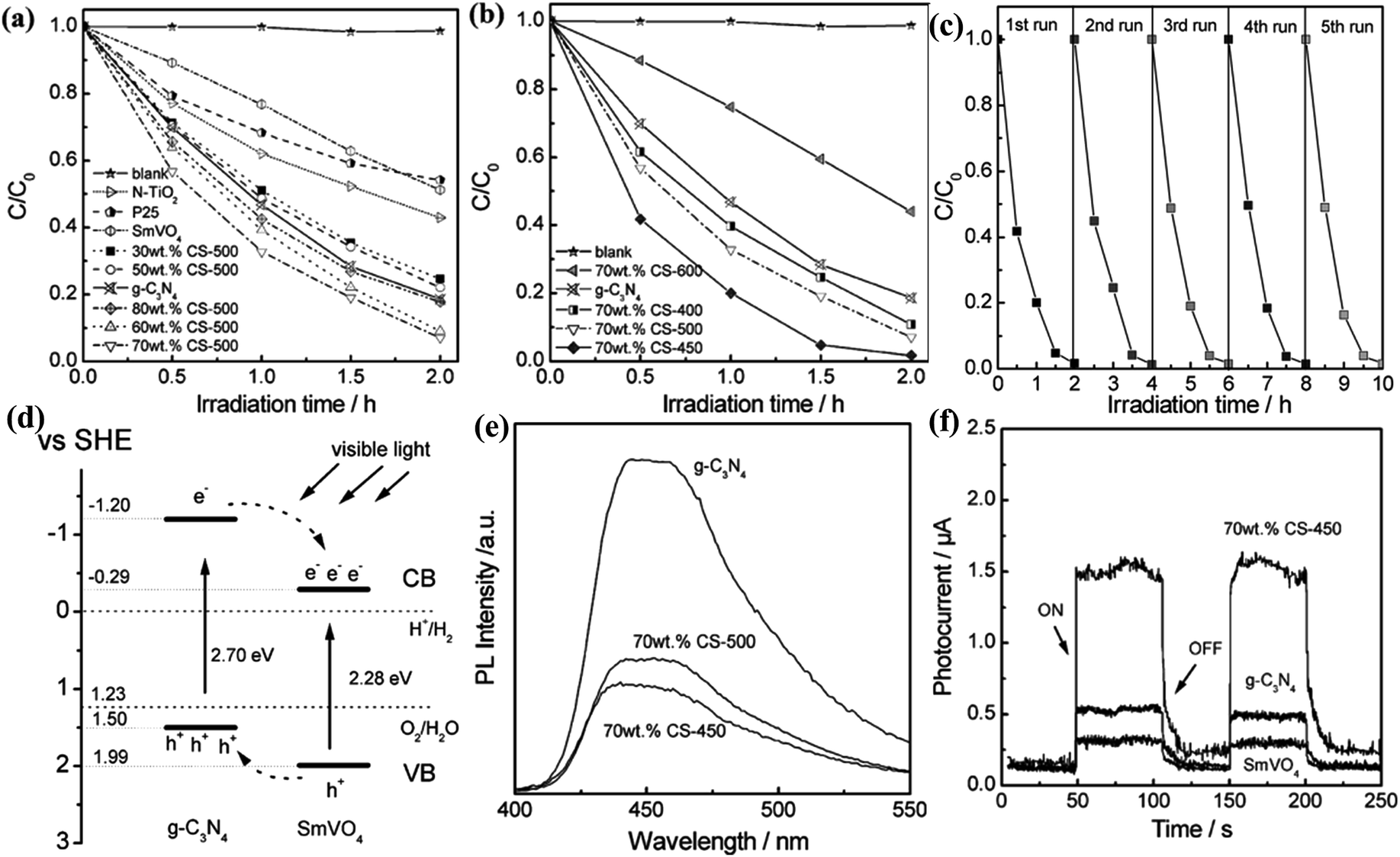 | ||
| Fig. 4 Visible light-driven photocatalytic activity of g-C3N4/SmVO4 composites with different g-C3N4 concentration (a) and 70 wt% g-C3N4/SmVO4 calcined at different temperatures (b). (c) Cycling runs for the photocatalytic degradation of RhB in the presence of 70 wt% CS-450 sample under visible light irradiation. (d) A schematic for electron–hole separation and transport at the visible light-driven g-C3N4/SmVO4 composite photocatalyst interface. (e) The photoluminescence spectra of pure g-C3N4 and g-C3N4/SmVO4 composite. (f) Transient photocurrent response for SmVO4, g-C3N4 and 70 wt% CS-450 samples. Reproduced with permission from ref. 35. | ||
3.3. Unique features of visible light-responsive photocatalysts
A photocatalysis process can be defined as a photo-induced reaction on the basis of semiconductor materials, which involves transforming light into other forms of energy. In a visible light-responsive photocatalytic system, three key features should be considered. The first is the electronic structure, which determines the optical properties and band structure of the photocatalysts. The semiconductor should have the ability to absorb visible light and the band-gap needs to be narrow enough to be excited by visible light photons. The second key feature is the crystallinity; high crystallinity means less crystalline defects, which would be beneficial for lowering the e−–h+ recombination rate. The final key feature is the surface character, which usually exerts a critical effect on surface chemical reactions such as oxidation by photo-generated holes and reduction by photo-generated electrons. The use of visible light-responsive photocatalysts to introduce suitable physicochemical properties is extremely common.Herein, another important point is that photo-absorption and e−–h+ generation are inextricably linked. Since the ability of a photocatalyst is exclusively governed by its band structure, from a thermodynamic point of view, estimation of the potential of the VB top and CB bottom is essential to predict the possibility of driving a photocatalytic reaction. A general way to carry out such estimation involves taking measurements of the optical absorption property, where the optical band-gap can be estimated using the following equation:
| Ahν = C(hν − Eg)n | (3.9) |
The photocatalytic removal efficiency of MO, RhB, and MB was enhanced under both simulated solar light and visible light irradiation when the photonic band-gaps of the TiO2 photonic crystals were well matched with the absorption peaks of the corresponding dyes. The improvement could be attributed to the intensified dye sensitization as a result of a slow photon effect on the edges of the photonic band-gaps.131 When commercial TiO2 crystals were coupled with a trace amount of narrow-band-gap Ag2CO3, the photocatalytic degradation activity under visible light irradiation of the composite was enhanced, due to the promoted visible light absorption and the suppressed recombination rate of the e−/h+ pairs.132 It was also demonstrated that the optical properties of the Cu2PO4OH hierarchical superstructures were strongly related to their morphologies and the size of the assembled crystallites, where the walnut-shaped morphology exhibited the best photocatalytic performance, due to its excellent visible light absorption ability, as well as high BET surface area.133
The redox potential of the reaction substrates, which adsorb on the surface depending on the amount of adsorbed substrates and on the surface chemical structure, depends more directly on the specific surface area. Table 1 summarizes the various preparation methods for visible light-responsive photocatalysts with various Brunauer–Emmett–Teller (BET) surface areas. In general, a high surface area to volume ratio of photocatalysts appears to be an important parameter for designing and engineering photocatalytic materials. For example, Dai et al.61 prepared C-doped Bi2O3 with a porous structure by a simply calcination of Bi(NO3)3·5H2O in glycol solution. The porous materials possess a larger specific surface area, which then contributed to more possible reaction sites on the photocatalyst surface, as well as facilitating the rapid diffusion of ions and molecules within the material, resulting in a higher photocatalytic activity. Kim et al.138 reported that the dye degradation performance was further markedly enhanced under visible light irradiation with a dramatic increase in the BET surface area, with an order of 2D BiOCl (5.5 m2 g−1) < 3D BiOCl (16.8 m2 g−1) ≪ BiOCl0.6I0.4 (46.6 m2 g−1). Such investigations indicate that the specific surface area plays an important role in the photo-degradation of pollutants. Moreover, the photocatalytic performances are not solely related to the surface area, but are also influenced by the surface charge, crystal facets, band structure, and optical properties, etc. For instance, owing to the change of surface charge and the newly created recombination center with Ag-loading, the photocatalytic degradation performance of Ag-doped BiOCl0.6I0.4 was dramatically reduced upon loading with small amounts of Ag, although the BET surface area increased from 46.6 m2 g−1 to 49.1 m2 g−1 with the Ag-loading. Our recent study on the sunlight photocatalytic performances of BiVO4 also found that the BET surface area was not the only factor influencing the photocatalytic activities.107
| Photocatalyst | Synthesis method | Optical properties and band-gap | BET surface area (m2 g−1) | Pollutant | Degradation efficiency or reaction rate | Crystalline phase | Ref. |
|---|---|---|---|---|---|---|---|
| WO3–TiO2/AC | Sol–gel | — | 323 | 10 mg L−1 Congo red | 62.52% | Anatase | 53 |
| 357 | 65.76% | ||||||
| 386 | 64.91% | ||||||
| 413 | 73.28% | ||||||
| 456 | 75.34% | ||||||
| 439 | 71.16% | ||||||
| 514 | 79.35% | ||||||
| 542 | 82.21% | ||||||
| 498 | 81.46% | ||||||
| C-doped Bi2O3 | Calcination | Visible light absorption band around 450–530 nm, the absorption edge of the C-doped Bi2O3 has an obvious red-shift compared with the pure Bi2O3 | 5.9 | 13 mg L−1 methyl orange | 95% | Monoclinic | 61 |
| Bi2O3 | 1.7 | 42% | |||||
| TiO2 | Hydrothermal | TiO2 nanotubes absorbed moderately around 385 nm and strongly around 300–350 nm, the optical absorption of Pt–TiO2 was enhanced significantly in the region of 300–700 nm. The band-gap of the TiO2 and Pt–TiO2 were calculated to be TiO2 (3.16 eV), 0.1 mol% Pt–TiO2 (3.11 eV), 0.5 mol% Pt–TiO2 (3.05 eV), 1 mol% Pt–TiO2 (3.01 eV), 2 mol% Pt–TiO2 (2.96 eV), 4 mol% Pt–TiO2 (2.64 eV) | 261.7 | Acetaldehyde | 0.0005 min−1 | Anatase | 66 |
| 0.1 mol% Pt–TiO2 | 226.7 | 0.0028 min−1 | |||||
| 0.5 mol% Pt–TiO2 | 203.9 | 0.0034 min−1 | |||||
| 1 mol% Pt–TiO2 | 163.6 | 0.0012 min−1 | |||||
| 2 mol% Pt–TiO2 | 141.8 | 0.0009 min−1 | |||||
| 4 mol% Pt–TiO2 | 139.9 | 0.0005 min−1 | |||||
| C–N–TiO2 (N(0)) | Microwave-assisted solvothermal reaction | Compared with those of commercial P25 powders, the absorption edges of the prepared samples apparently shifted to the visible range. The band-gap energies deduced from the tangent line are 3.16, 3.11, 2.98, 2.85, and 2.77 eV for P25, N(0), DEA, TMA, and DETA, respectively | 59.0 | 5 mg L−1 RhB | 48%, 0.0049 min−1 | Anatase | 69 |
| C–N–TiO2 (DEA) | 69.7 | 59%, 0.0142 min−1 | |||||
| C–N–TiO2 (TMA) | 63.7 | 92%, 0.0397 min−1 | |||||
| C–N–TiO2 (DETA) | 129.1 | 10%, 0.0017 min−1 | |||||
| TiO2 | Acid catalysed sol–gel | TiO2 showed an absorption threshold at 406 nm and a band-gap energy of 3.05 eV; N–TiO2 showed a slight red-shift, giving a band-gap of 3.02 eV; N–Pt–TiO2 had the longest absorption edge and the highest absorbance in the visible light region, giving a band-gap of 2.58 eV | 46.6 | 20 mg L−1 phenol | 35.6% | Anatase | 70 |
| N–TiO2 | 78.5 | 41.9% | |||||
| N–Pt–TiO2 | 94.5 | 100% | |||||
| ZnO | Hybridized interaction | Both ZnO and C60–ZnO showed the absorbance edge from 400 nm to 800 nm, the absorption intensity changed with the increasing of the C60 amount | 57.3 | 3 mg L−1 MB | 85%, 0.0337 min−1 | Hexagonal | 86 |
| C60–ZnO | 56.9 | 95%, 0.0569 min−1 | |||||
| 4.9 wt% g-C3N4–ZnO | Calcination | The absorption edges of g-C3N4–ZnO composite samples shift significantly to longer wavelengths compared to ZnO, as well as the band-gap narrowing | 36 | 3 mg L−1 methyl orange or p-nitrophenol | 15.6 wt% g-C3N4–ZnO exhibited the best photocatalytic performance | Both ZnO and g-C3N4 phases | 96 |
| 8.4 wt% g-C3N4–ZnO | 75 | ||||||
| 15.6 wt% g-C3N4–ZnO | 33 | ||||||
| 58.1 wt% g-C3N4–ZnO | 28 | ||||||
| A-BiVO4 (pH = 4.9) | Hydrothermal | The absorption edges for both the A-BiVO4 and S-BiVO4 samples blue shift eventually as the pH values of the precursors increase. As for A-BiVO4, the absorption edge is measured to be at approx. 560 nm (pH 4.9), 555 nm (pH 6.26), and 540 nm (pH 7); as 590 nm (pH 4.9), 540 nm (pH 6.26), and 525 nm (pH 7) for S-BiVO4. The estimated band-gap energies of A-BiVO4 were approx. 2.40 eV (pH 4.9), 2.43 eV (pH 6.26), and 2.45 eV (pH 7), respectively, whereas for S-BiVO4, the band-gap energies were measured to be approx. 2.28 eV (pH 4.9), 2.40 eV (pH 6.26), and 2.45 eV (pH 7) | 10.3 | 5 mg L−1 RhB | 61% | Monoclinic scheelite | 107 |
| A-BiVO4 (pH = 6.26) | 4.6 | 89% | |||||
| A-BiVO4 (pH = 7) | 1.6 | 62% | |||||
| S-BiVO4 (pH = 4.9) | 6.3 | 97% | |||||
| S-BiVO4 (pH = 6.26) | 4.6 | 96% | |||||
| S-BiVO4 (pH = 7) | 11.1 | 61% |
Apart from the crystal facet and electronic structures, the existence of surface defects on the photocatalysts remains another important factor affecting its photocatalytic performance. The roles the defects play in the adsorption and surface reactivity have been acknowledged and extensively characterized by various techniques.132,139,140 Yu et al.132 revealed that more surface hydroxyl groups over the Ag2CO3/TiO2 composite could react with more photo-generated h+ and produce more ˙OH radicals to decompose the dye. Zhang et al.139 demonstrated that the visible hydroxyl groups indicated the existence of surface defects on ZnO nanorods, and that the existence of surface defects played a positive role in the photocatalytic activity of ZnO nanorods; whereby, the photo-generated holes could be trapped by surface defects and the separation of photo-generated e−/h+ pairs was facilitated. Moreover, the photo-generated holes trapped by surface defects more readily reacted with electron donors and the photocatalytic reaction could thus be greatly promoted; this was the reason why ZnO nanorods with surface defects showed significantly higher photocatalytic performances. Bai et al.140 showed that ZnO1−x with surface oxygen defects could be excited by visible light, due to the narrowing energy band-gap resulting from the generation of the surface defect level induced by the surface oxygen-defect states. The surface defects may serve as adsorption sites as well as charge carrier traps, where the charge transfers to the adsorbed species and prevents the e−/h+ recombination. However, TaON nanoparticles with lower surface reduction defect sites exhibited enhanced photocatalytic performance for the mineralization of phenol and its chloro-derivatives in an aqueous phase under visible light irradiation.37 A possible interpretation of this phenomenon is that the photocatalytic performance may not be governed by a single feature. Therefore, in a diverse photocatalysis system, combined photocatalysts in different combinations and ratios could provide varied results depending upon the conditions used.
4. Application of visible light-responsive photocatalysts in water treatment
Visible light-responsive photocatalysts are expected to play an important role in tackling growing global concerns over water contamination, which is of great significance in alleviating the increasingly serious global water resource crisis. The photocatalysis route itself holds many advantages, such as strong oxidation power, moderate operation temperature, and green-chemistry related procedures, offering a tantalizing route to meet the global challenges associated with the environment, energy, and sustainability, and with the aid of abundant sunlight resources. Visible light-responsive photocatalysts have been widely used for the treatment of inorganic, organic, and biological contaminated water. In this section, the main applications of visible light-responsive photocatalysts in water treatment are briefly summarized, but it is worth noting that most applications discussed here are still in the stage of laboratory research.4.1. Photocatalytic degradation of organic pollutants
In most cases, different types of dyes are studied as model compounds for the photocatalytic degradation of large organic molecules in water treatment. Organic dyes are often used in textile, printing, and photographic industries, but a sizable fraction of dyes is wasted in the dying process and is released into effluent water streams. In general, the presence of even low concentrations of dyes in effluent streams seriously affects the nature of water, and is difficult to be biodegraded or oxidized with the aid of chemicals. Therefore, appreciable efforts have been made to develop photocatalysts to degrade dyes in aqueous solutions under visible light.141–143 Bismuth titanate (Bi12TiO20) nanostructures with different morphologies have been used to photocatalytically remove acid orange 7 under visible light irradiation, with tests showing that Bi12TiO20 has a higher photocatalytic activity than that of traditional N-doped TiO2; in this regard, the BET surface area plays an important role in the reaction efficiency, while the crystallinity of the samples is another important factor that could also boost the photocatalytic performances.51 The photosensitization of photocatalysts by dyes has also been used for the visible photo-degradation and mineralization of dye pollutants. Li et al.141 initiated a comparison study of dye photodegradation over TiO2 and ZnO, where the microscale ZnO exhibited much higher visible photocatalytic activity than that of P25, due to the higher photosensitization efficiency of electron transfer from an excited dye to the CB of the microscale ZnO than that of P25. The better crystallinity and lower defects of the microscale ZnO than that of the nanosized ZnO resulted in a better photostability. TiO2-modified ZnO was developed as a novel light-to-electricity conversion device, and has proven to be a promising candidate for the photocatalytic removal of dye pollutants, as well as a renewable energy source.Pharmaceutical and personal care products (PPCPs) have recently been considered as emerging contaminants, and are an extraordinarily diverse group of chemicals used in prescription and nonprescription drugs, human health and cosmetic care, veterinary medicine, and agricultural practice.144 Specific PPCPs may cause ecological harm, such as endocrine disruption and antimicrobial resistance, thus some of PPCPs have been classified as “priority pollutants” by both the US Environmental Protection Agency and the European Union Water Framework Directive. PPCPs have frequently been studied with respect to environmental protection because of their toxicity and nonbiodegradability. To date, visible light photocatalytic degradation has been considered as a promising approach for the environmentally friendly decomposition of PPCPs, offering high efficiency, cost effectiveness, ease of operation, etc. For example, An and Zhou145 employed a new combined catalyst copper-plating iron-doped Cu2O (FeCu/Cu2O) to degrade a mixture of five commonly used PPCPs (sulfamethoxazole, oxytetracyclin, paracetamol, aspirin, and triclosan) under visible light irradiation. Compared with the Fe/C inner micro-circuit, the electric currents flowing between Cu and Fe increased the speed of anodic Fe dissolution. It was found that Cu2O could accelerate the PPCP degradation processes under visible light irradiation, due to its photochemical properties. Moreover, the increased dissolved oxygen concentration in the solution by shaking not only preconditioned the photo-catalysis reaction, but also set the stage for Fe reduction. Zhao et al.146 investigated the photochemical degradation of the antibiotic oxytetracycline (OTC) with nitrogen- and fluorine-doped titanium dioxide (NF–TiO2) film at different pH values in aqueous solutions under visible and solar light irradiation. The kinetics and mechanism during the photolytic and photocatalytic degradation of OTC were intensively studied. The photochemical degradation of OTC with NF–TiO2 film could occur via a number of competing reaction processes, such as direct photolytic degradation, as shown in Fig. 5; whereby, the electrons and holes could be separated by the excited NF–TiO2 under light irradiation, followed by the fact that a series of active redox species were produced by a series of reactions, leading to the oxidative-reductive degradation of OTC.
 | ||
| Fig. 5 Influence of TBA (10 mM), NaN3 (5 mM), KI (10 mM) and catalase (6 unit per L) on the photocatalytic degradation of OTC by NF–TiO2 under visible light at pH 5.5 (a) and 8.5 (b) (OTC 5 mg L−1). (c) The initial rate of OTC photolytic degradation per unit of concentration at various initial concentrations at pH 5.5 and 8.5 under visible and solar light. (d) The proposed pathway of OTC photolytic degradation. Reproduced with permission from ref. 146. | ||
Phenolic compounds can cause various diseases, including cancer, angiocardiopathy, and gastroenterology etc., even at very low concentrations, and represent a typical family of organic pollutants widely present in wastewater from petrol, coal, and other chemical industries.147 Most phenolic compounds are usually difficult to be mineralized by a biodegradation method, due to the stable benzene ring and its recalcitrant nature. However, such compounds have been reported to be effectively degraded by visible light-responsive photocatalysts.30,37,79,127,147 Li and colleagues147 synthesized a novel layered perovskite crystal La2NiO4 photocatalyst with high activity for mineralizing 4-chlorophenol under visible light irradiation. First, the 4-chlorophenol was ionized into anions and donated electrons to La2NiO4 due to its positively charged surface. Then, the electrons could react with dissolved O2 to produce ˙O2− radicals, followed by reacting with H+ to form ˙OH radicals, which could oxidize 4-chlorophenol into CO2, thus leading to complete degradation. Other organic pollutions, such as benzyl alcohol,148 methanol,149 benzyl amine,150 hydroxytyrosol,151 and benzene,152 in aqueous solutions have also been reported to be efficiently degraded by visible light induced photocatalytic.
4.2. Photocatalytic degradation of inorganic pollutants
The presence of inorganic impurities, such as residual ions and acids, in the water matrix has distinctive effects toward a water system's ecological environment. Some of them are highly toxic to most of living organisms when their concentration levels are higher than a certain value. For instance, hexavalent chromium (Cr(VI)) is a carcinogenic and mutagenic pollutant, which is frequently found in wastewater, possibly stemming from pigment production, metal plating, and leather tanning, etc. The visible light induced photocatalytic reduction of aqueous Cr(VI) has received much attention recently, due to its low cost and high efficiency without secondary pollution.153,154 For instance, synthesized SnIn4S8 particles with a flower-like nanostructure could exhibit an excellent photocatalytic reduction efficiency of aqueous Cr(VI) (∼97%) and good photocatalytic stability.153 The strong absorption in the visible light region, large surface area, and excellent charge separation characteristics of SnIn4S8 are responsible for its promising removal performance. The mechanism for Cr(VI) removal from water by SnIn4S8 is shown in Fig. 6. The photo-generated electrons in the VB of SnIn4S8 could be excited to the CB, while holes are then generated in the VB. Subsequently, a portion of the photo-generated e−–h+ pairs migrate to the surface of the SnIn4S8 and participate in the redox reaction. The photo-generated e− reduces the adsorbed Cr(VI) to Cr(III), while the h+ enables the oxidization of water to O2. By this means, the photocatalytic reduction of Cr(VI) coupled with the synergistic effect of photo-degradation of bisphenol A in aqueous solution is also achieved by using Bi2O3/TiO2 under visible light irradiation.154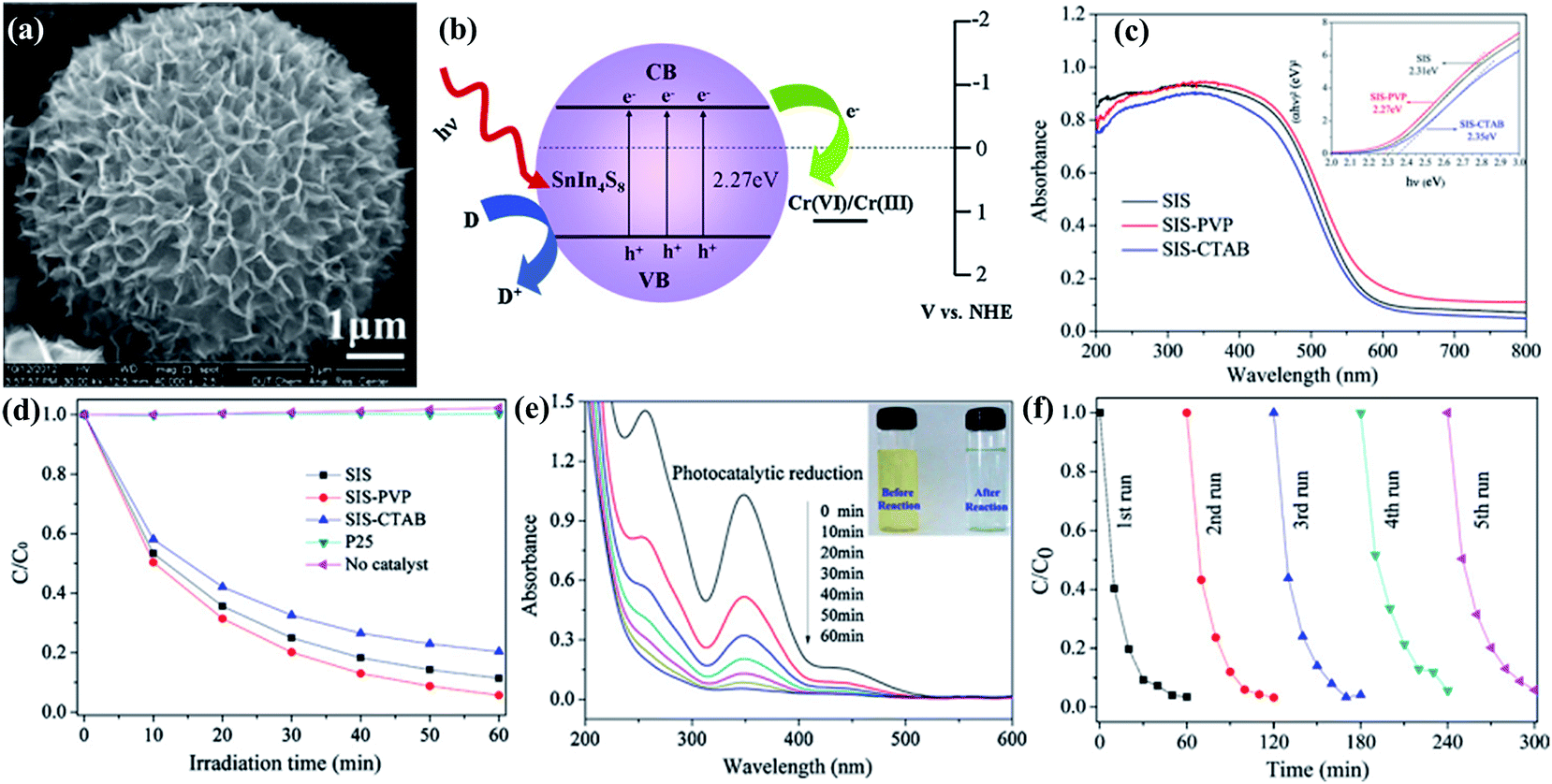 | ||
| Fig. 6 (a) SEM image of SnIn4S8 nanostructure. (b) Schematic diagram for the photocatalytic reduction of Cr(VI) ions by SnIn4S8 under visible light irradiation. (c) UV-vis diffuse reflectance spectra of the samples SIS, SIS-PVP and SIS-CTAB. Inset: plots of (Ahν)2 versus hν for calculating the band-gap energy. (d) Comparison of the photocatalytic reduction of aqueous Cr(VI) over the as-prepared samples and the commercial P25 TiO2 under visible light irradiation. (e) UV-vis absorbance spectrum of K2Cr2O7 solution in the presence of SIS-PVP products under visible light irradiation. The inset shows the digital images of the solution before and after reaction. (f) The photocatalytic reduction of aqueous Cr(VI) over the sample SIS-PVP with five times cycling use. Reproduced with permission from ref. 153. | ||
4.3. Photocatalytic disinfection of biological pollutants
The different types of microbes (bacteria, viruses, fungi, algae, plankton, etc.) present in wastewater are harmful to health and can cause various diseases.155 Over the past few decades, the visible light activated photocatalytic disinfection of water has received significant attention, with research focus moving from laboratory studies to potential applications.156,157 For example, a dye-sensitized TiO2 thin film was used in the photocatalytic disinfection of phytopathogenic bacteria under visible light irradiation. The inhibition rates of Erwinia carotovora subsp. carotovora 3, Enterobacter cloacae SM1, and E. carotovora subsp. carotovora 7 that could induce severe soft/basal rot disease in vegetable crops were higher than 90%. These results indicated that the visible light-responsive dye-sensitized TiO2 thin film had the potential for direct application to plant protection in water systems.156 A visible light-activated palladium-modified nitrogen-doped titanium oxide (TiON/PdO) photocatalyst has been demonstrated with good visible light adsorption and a superior efficient photocatalytic disinfection effect on Fungi Fusarium graminearum macroconidia. The disinfection effect benefitted from the strong adsorption of the TiON/PdO photocatalyst onto the Fusarium graminearum macroconidia surface due to their opposite surface charges. The photocatalytic disinfection mechanism of the TiON/PdO photocatalyst on the Fusarium graminearum macroconidia is displayed in Fig. 7, and is attributed to their cell wall/membrane damage caused by the attack from the reactive oxygen species (ROSs), while a breakage of their cell structure was not necessary for their loss of viability.157 | ||
| Fig. 7 Photocatalytic disinfection mechanism of TiON/PdO photocatalyst on Fusarium graminearum macroconidia under visible light irradiation. Reproduced with permission from ref. 157, copyright (2013) American Chemical Society. | ||
5. Conclusion and perspectives
Visible light-responsive photocatalysis for water and wastewater treatment is gaining momentum globally and presenting great opportunities to revolutionize water and wastewater treatment due to its unique features. In this review, we have extensively reviewed the current progress in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts. The fundamentals of heterogeneous photocatalysis and the regulatory mechanism of visible light-responsive photocatalysis, as well as the unique features of visible light-responsive photocatalysts have been briefly discussed. To extend the optical response of photocatalysts into the visible region, a variety of methods, such as the modification of traditional photocatalysts via doping, dye sensitization, formation of a heterostructure, coupled with π-conjugated structure, and exploration of novel multi-component oxides and nanocomposites with visible light responses, have been developed. These photocatalysts have been widely used for the degradation of inorganic and organic pollutants, as well as for photocatalytic disinfection. Therefore, these results show promise for the further development of sustainable environmental remediation technologies based on photocatalytic redox reactions driven by visible light as a renewable source of energy.Although steady progress in heterogeneous photocatalytic water treatment using visible light-responsive photocatalysts has been achieved,158 studies in this field are still at the immature stage and further developments are required. The challenges faced in developing water treatment with visible light-responsive photocatalysts are significant, but many are perhaps only temporary including the high cost, technical hurdles, and potential environmental and human risks. To promote the feasibility of visible light-responsive photocatalytic water treatment technology in the near future, several key technical constraints, ranging from catalyst development to reactor design and process optimization, have to be addressed including the following specific aspects:
(i) Improving the efficiency and photo-stability of the visible light-responsive photocatalysts. The performance of visible light-responsive photocatalysts is currently limited by the physicochemical properties of these materials. For example, among the modification methods, though dye sensitization has been able to extend the adsorption light wavelength to the visible light range and improve the activity of traditional photocatalysts under visible light, the usage of such photocatalytic materials is still limited due to issues regarding the dissolution and degradation of dyes, which retards their photocatalysis applications. Therefore, a more careful design of the functional photocatalyst is required to obtain suitable physicochemical properties of the materials.
(ii) Devising an appropriate photocatalyst immobilization strategy to provide a cost-effective solid–liquid separation. One detrimental limitation at the current stage is running-out of the catalyst during the photocatalytic process, which jeopardizes the regeneration of the catalysts and poses adverse impacts on the environment due to the leakage of the photocatalyst. Immobilized photocatalytic systems can avoid problems associated with catalyst recovery and agglomeration, as well as minimizing the scale of the reactor.
(iii) Designing an effective photoreactor for the full utilization of solar energy to reduce the electricity costs. The implementation of photocatalytic processes at an industrial level requires the design of suitable photoreactors, where a proper design and build-up of such devices would be helpful to better harvest solar energy, accommodate the photocatalysts and reactants, as well as collect the reaction products.
(iv) Establishing a globally experimental database regarding the information of the photocatalysts tested including the types of catalyst materials, preparation routes, modifications, photocatalytic reaction environments and activity. Such a database should be easily accessible to avoid any repeated, unnecessary work and should guide the development of innovative catalysts. To obtain the desired photo-degradation efficiency, combining different techniques and approaches might always be indispensable. In this regard, a database detailing the fabrication and usage of the existing photocatalysts needs to be established because plenty of nanostructured materials have already been used in visible light-responsive photocatalysis. The accumulation of a large amount of theoretical and modelling work is also useful and imperative in the quest to foster a deep understanding of the preparation, properties and performances of photocatalysts and their optimization for water treatment. Multi-technology integration will provide a bright prospect for water treatment and energy-related issues using visible light-responsive photocatalysts with advanced efficiency and good robustness.
Acknowledgements
The authors are grateful for the financial support from the Key Science and Technology Program of Henan Province, PR China (Grant nos 122102310486 and 132102210129), the Basic Scientific and Technological Frontier Project of Henan Province, PR China (Grant nos 102300410098, 132330410138, and 122300410293). The authors also would like to thank the Innovation Scientists and Technicians Troop Construction Projects of Henan Province, the Plan for Scientific Innovation Talent of Henan Province (Grant no. 134200510014), the Fostering Foundation of Henan Normal University for the Author of National Excellent Ph.D. Dissertation, PR China (Grant no. 01333900011), and the National Natural Science Foundation of China (Grants no. 21477034).References
- J. Qu and M. Fan, Crit. Rev. Environ. Sci. Technol., 2010, 40, 519–560 CrossRef CAS.
- X. Zhou, Y. Li and Y. Zhao, RSC Adv., 2014, 30, 15620–15629 RSC.
- V. K. Gupta, I. Ali, T. A. Saleh, A. Nayak and S. Agarwal, RSC Adv., 2012, 2, 6380–6388 RSC.
- J. Sun, B. Zhang, R. Sun, Y. Li and J. Wu, Int. J. Environ. Pollut., 2009, 38, 81–87 CrossRef CAS.
- S. Sun, C. Li, J. Sun, S. Shi, M. Fan and Q. Zhou, J. Hazard. Mater., 2009, 161, 1052–1057 CrossRef CAS PubMed.
- J. Sun, S. Sun, M. Fan, H. Guo, Y. Lee and R. Sun, J. Hazard. Mater., 2008, 153, 187–193 CrossRef CAS PubMed.
- O. Ganzenko, D. Huguenot, E. D. van Hullebusch, G. Esposito and M. A. Oturan, Environ. Sci. Pollut. Res., 2014, 21, 8493–8524 CrossRef CAS PubMed.
- S. Ray, M. Takafuji and H. Ihara, RSC Adv., 2013, 3, 23664–23672 RSC.
- M. Tanveer and G. T. Guyer, Renewable Sustainable Energy Rev., 2013, 24, 534–543 CrossRef CAS PubMed.
- L. Yang, S. Dong, J. Sun, J. Feng, Q. Wu and S. Sun, J. Hazard. Mater., 2010, 179, 438–443 CrossRef CAS PubMed.
- P. Fageria, S. Gangopadhyay and S. Pande, RSC Adv., 2014, 4, 24962–24972 RSC.
- S. Balachandran, S. G. Praveen, R. Velmurugan and M. Swaminathan, RSC Adv., 2014, 4, 4353–4362 RSC.
- Y. C. Chang, RSC Adv., 2014, 4, 20273–20280 RSC.
- L. G. Devi and R. Kavitha, Appl. Catal., B, 2013, 140, 559–587 CrossRef PubMed.
- D. Kanakaraju, B. D. Glass and M. Oelgemoeller, Environ. Chem. Lett., 2014, 12, 27–47 CrossRef CAS PubMed.
- A. Rey, P. Garcia-Munoz, M. D. Hernandez-Alonso, E. Mena, S. Garcia-Rodriguez and F. J. Beltran, Appl. Catal., B, 2014, 154, 274–284 CrossRef PubMed.
- J. Sun, L. Qiao, S. Sun and G. Wang, J. Hazard. Mater., 2008, 155, 312–319 CrossRef CAS PubMed.
- Z. Li, Y. Fang and S. Xu, Mater. Lett., 2013, 93, 345–348 CrossRef CAS PubMed.
- L. Z. Qin, H. Liang, B. Liao, A. D. Liu, X. Y. Wu and J. Sun, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 307, 385–390 CrossRef CAS PubMed.
- Y. Min, K. Zhang, Y. Chen and Y. Zhang, Chem. Eng. J., 2011, 175, 76–83 CrossRef CAS PubMed.
- Q. Li, L. Zong, Y. Xing, X. Wang, L. Yu and J. Yang, Sci. Adv. Mater., 2013, 5, 1316–1322 CrossRef CAS PubMed.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS PubMed.
- C. Mondal, M. Ganguly, J. Pal, A. Roy, J. Jana and T. Pal, Langmuir, 2014, 30, 4157–4164 CrossRef CAS PubMed.
- W. Wang, J. Wang, Z. Wang, X. Wei, L. Liu, Q. Ren, W. Gao, Y. Liang and H. Shi, Dalton Trans., 2014, 43, 6735–6743 RSC.
- S. Zhang, Ceram. Int., 2014, 40, 4553–4557 CrossRef CAS PubMed.
- Z. Li, S. Yang, J. Zhou, D. Li, X. Zhou, C. Ge and Y. Fang, Chem. Eng. J., 2014, 241, 344–351 CrossRef CAS PubMed.
- S. Wang, D. Li, C. Sun, S. Yang, Y. Guan and H. He, J. Mol. Catal. A: Chem., 2014, 383, 128–136 Search PubMed.
- H. Huang, Y. Feng, J. Zhou, G. Li and K. Dai, Desalin. Water Treat., 2013, 51, 7236–7240 CrossRef CAS.
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- J. Sheng, X. Li and Y. Xu, ACS Catal., 2014, 4, 732–737 CrossRef CAS.
- Y. S. Xu, Y. X. Yu and W. D. Zhang, J. Nanosci. Nanotechnol., 2014, 14, 6800–6808 CrossRef CAS PubMed.
- U. M. Garcia-Perez, A. Martinez-de la Cruz and J. Peral, Electrochim. Acta, 2012, 81, 227–232 CrossRef CAS PubMed.
- S. Dong, J. Sun, Y. Li, C. Yu, Y. Li and J. Sun, Appl. Catal., B, 2014, 144, 386–393 CrossRef CAS PubMed.
- M. Shang, W. Wang, L. Zhou, S. Sun and W. Yin, J. Hazard. Mater., 2009, 172, 338–344 CrossRef CAS PubMed.
- T. Li, L. Zhao, Y. He, J. Cai, M. Luo and J. Lin, Appl. Catal., B, 2013, 129, 255–263 CrossRef CAS PubMed.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maccato, C. Maragno, E. Tondello, U. L. Stangar, M. Bergant and D. Mahne, Nanotechnology, 2007, 18, 375709 CrossRef.
- Y. Chen, S. Liang, L. Wen, W. Wu, R. Yuan, X. Wang and L. Wu, Phys. Chem. Chem. Phys., 2013, 15, 12742–12747 RSC.
- D. Barreca, A. P. Ferrucci, A. Gasparotto, C. Maccato, C. Maragno and E. Tondello, Chem. Vap. Deposition, 2007, 13, 618–625 CrossRef CAS.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maccato, E. Tondello, O. I. Lebedev, S. Turner, G. Van Tendeloo, C. Sada and U. L. Stangar, ChemPhysChem, 2009, 10, 3249–3259 CrossRef CAS PubMed.
- G. Carraro, R. Sugrañez, C. Maccato, A. Gasparotto, D. Barreca, C. Sada, M. Cruz-Yusta and L. Sánchez, Thin Solid Films, 2014, 564, 121–127 CrossRef CAS PubMed.
- A. Abd Aziz, C. K. Cheng, S. Ibrahim, M. Matheswaran and P. Saravanan, Chem. Eng. J., 2012, 183, 349–356 CrossRef CAS PubMed.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- E. Casbeer, V. K. Sharma and X. Z. Li, Sep. Purif. Technol., 2012, 87, 1–14 CrossRef CAS PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS PubMed.
- K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2010, 12, 14420–14432 RSC.
- X. Qu, P. J. J. Alvarez and Q. Li, Water Res., 2013, 47, 3931–3946 CrossRef CAS PubMed.
- X. G. Yan, L. Xu, W. Q. Huang, G. F. Huang, Z. M. Yang, S. Q. Zhan and J. P. Long, Mater. Sci. Semicond. Process., 2014, 23, 34–41 CrossRef CAS PubMed.
- L. Gu, J. Wang, Z. Zou and X. Han, J. Hazard. Mater., 2014, 268, 216–223 CrossRef CAS PubMed.
- X. Zhu, J. Zhang and F. Chen, Chemosphere, 2010, 78, 1350–1355 CrossRef CAS PubMed.
- J. Sun, S. Dong, Y. Wang and S. Sun, J. Hazard. Mater., 2009, 172, 1520–1526 CrossRef CAS PubMed.
- J. Sun, Y. Wang, R. Sun and S. Dong, Mater. Chem. Phys., 2009, 115, 303–308 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- C. Di Valentin and G. Pacchioni, Catal. Today, 2013, 206, 12–18 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, Nanoscale, 2014, 6, 1946–2008 RSC.
- Y. Wang, C. Feng, M. Zhang, J. Yang and Z. Zhang, Appl. Catal., B, 2010, 100, 84–90 CrossRef CAS PubMed.
- B. Neumann, P. Bogdanoff, H. Tributsch, S. Sakthivel and H. Kisch, J. Phys. Chem. B, 2005, 109, 16579–16586 CrossRef CAS PubMed.
- Y. J. Chen, G. Y. Jhan, G. L. Cai, C. S. Lin, M. S. Wong, S. C. Ke, H. H. Lo, C. L. Cheng and J. J. Shyue, J. Vac. Sci. Technol., A, 2010, 28, 779–782 CAS.
- G. Dai, S. Liu and Y. Liang, J. Alloys Compd., 2014, 608, 44–48 CrossRef CAS PubMed.
- M. Samadi, H. A. Shivaee, M. Zanetti, A. Pourjavadi and A. Moshfegh, J. Mol. Catal. A: Chem., 2012, 359, 42–48 CrossRef CAS PubMed.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- J. Li, J. Xu and J. Huang, CrystEngComm, 2014, 16, 375–384 RSC.
- B. K. Vijayan, N. M. Dimitrijevic, J. Wu and K. A. Gray, J. Phys. Chem. C, 2010, 114, 21262–21269 CAS.
- J. Sun, S. Dong, J. Feng, X. Yin and X. Zhao, J. Mol. Catal. A: Chem., 2011, 335, 145–150 CrossRef CAS PubMed.
- H. F. Moafi, M. A. Zanjanchi and A. F. Shojaie, J. Nanosci. Nanotechnol., 2014, 14, 7139–7150 CrossRef CAS PubMed.
- Y. C. Wu and L. S. Ju, J. Alloys Compd., 2014, 604, 164–170 CrossRef CAS PubMed.
- H. Sun, G. Zhou, S. Liu, H. M. Ang, M. O. Tade and S. Wang, Chem. Eng. J., 2013, 231, 18–25 CrossRef CAS PubMed.
- J. Zhao, C. Chen and W. Ma, Top. Catal., 2005, 35, 269–278 CrossRef CAS PubMed.
- Z. Li, Y. Fang, X. Zhan and S. Xu, J. Alloys Compd., 2013, 564, 138–142 CrossRef CAS PubMed.
- D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef CAS PubMed.
- J. Shang, F. Zhao, T. Zhu and J. Li, Sci. China: Chem., 2011, 54, 167–172 CrossRef CAS.
- X. Li, Y. Cheng, S. Kang and J. Mu, Appl. Surf. Sci., 2010, 256, 6705–6709 CrossRef CAS PubMed.
- G. C. C. Yang and S. W. Chan, J. Nanopart. Res., 2009, 11, 221–230 CrossRef CAS.
- J. Li, Z. Guo, Y. Wang and Z. Zhu, Micro Nano Lett., 2014, 9, 65–68 Search PubMed.
- Y. Wu, F. Xu, D. Guo, Z. Gao, D. Wu and K. Jiang, Appl. Surf. Sci., 2013, 274, 39–44 CrossRef CAS PubMed.
- W. Teng, X. Li, Q. Zhao and G. Chen, J. Mater.Chem. A, 2013, 1, 9060–9068 CAS.
- M. Lee and K. Yong, Nanotechnology, 2012, 23, 194014 CrossRef PubMed.
- Z. Wei, Y. Li, S. Luo, C. Liu, D. Meng, M. Ding and G. Zeng, Sep. Purif. Technol., 2014, 122, 60–66 CrossRef CAS PubMed.
- W. Zhou, Y. Guan, D. Wang, X. Zhang, D. Liu, H. Jiang, J. Wang, X. Liu, H. Liu and S. Chen, Chem.–Asian J., 2014, 9, 1648–1654 CrossRef CAS PubMed.
- Y. Ni, W. Wang, W. Huang, C. Lu and Z. Xu, J. Colloid Interface Sci., 2014, 428, 162–169 CrossRef CAS PubMed.
- R. T. Thomas, P. Abdul Rasheed and N. Sandhyarani, J. Colloid Interface Sci., 2014, 428, 214–221 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- H. Fu, T. Xu, S. Zhu and Y. Zhu, Environ. Sci. Technol., 2008, 42, 8064–8069 CrossRef CAS.
- Y. Long, Y. Lu, Y. Huang, Y. Peng, Y. Lu, S. Z. Kang and J. Mu, J. Phys. Chem. C, 2009, 113, 13899–13905 CAS.
- D. Chen, K. Wang, D. Xiang, R. Zong, W. Yao and Y. Zhu, Appl. Catal., B, 2014, 147, 554–561 CrossRef CAS PubMed.
- F. Wang, S. Min, Y. Han and L. Feng, Superlattices Microstruct., 2010, 48, 170–180 CrossRef CAS PubMed.
- X. Li, D. Wang, G. Cheng, Q. Luo, J. An and Y. Wang, Appl. Catal., B, 2008, 81, 267–273 CrossRef CAS PubMed.
- F. Deng, L. Min, X. Luo, S. Wu and S. Luo, Nanoscale, 2013, 5, 8703–8710 RSC.
- Y. Min, K. Zhang, W. Zhao, F. Zheng, Y. Chen and Y. Zhang, Chem. Eng. J., 2012, 193–194, 203–210 CrossRef CAS PubMed.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, J. Appl. Phys., 2014, 115 Search PubMed.
- S. Dong, Y. Li, J. Sun, C. Yu, Y. Li and J. Sun, Mater. Chem. Phys., 2014, 145, 357–365 CrossRef CAS PubMed.
- T. Hasobe, S. Hattori, P. V. Kamat and S. Fukuzumi, Tetrahedron, 2006, 62, 1937–1946 CrossRef CAS PubMed.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756 RSC.
- M. Fu, J. Liao, F. Dong, H. Li and H. Liu, J. Nanomater., 2014, 2014, 1–8 Search PubMed.
- Q. Luo, L. Bao, D. Wang, X. Li and J. An, J. Phys. Chem. C, 2012, 116, 25806–25815 CAS.
- F. F. Li, D. F. Yang, M. S. Xia, Y. Wang and Y. S. Jiang, J. Inorg. Mater., 2011, 26, 917–922 CrossRef CAS.
- J. Y. Zheng, S. H. Bao, Y. Guo and P. Jin, ACS Appl. Mater. Interfaces, 2014, 6, 5940–5946 CAS.
- G. Chen, M. Sun, Q. Wei, Y. Zhang, B. Zhu and B. Du, J. Hazard. Mater., 2013, 244, 86–93 CrossRef PubMed.
- Y. C. Nah, I. Paramasivam and P. Schmuki, ChemPhysChem, 2010, 11, 2698–2713 CrossRef CAS PubMed.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Su, Y. Zhang, S. Xu, S. Wang, H. Ding, S. Pan, G. Wang, G. Li and H. Zhao, Nanoscale, 2014, 6, 5181–5192 RSC.
- F. Duan, Q. Zhang, Q. Wei, D. Shi and M. Chen, Prog. Chem., 2014, 26, 30–40 Search PubMed.
- P. Madhusudan, J. Yu, W. Wang, B. Cheng and G. Liu, Dalton Trans., 2012, 41, 14345–14353 RSC.
- S. Dong, J. Feng, Y. Li, L. Hu, M. Liu, Y. Wang, Y. Pi, J. Sun and J. Sun, Appl. Catal., B, 2014, 152, 413–424 CrossRef PubMed.
- S. Dong, C. Yu, Y. Li, Y. Li, J. Sun and X. Geng, J. Solid State Chem., 2014, 211, 176–183 CrossRef CAS PubMed.
- C. Yu, S. Dong, J. Feng, J. Sun, L. Hu, Y. Li and J. Sun, Environ. Sci. Pollut. Res., 2014, 21, 2837–2845 CrossRef CAS PubMed.
- L. Hu, S. Dong, Y. Li, Y. Pi, J. Wang, Y. Wang and J. Sun, J. Taiwan Inst. Chem. Eng., 2014, 45, 2462–2468 CrossRef CAS PubMed.
- X. J. Dai, Y. S. Luo, W. D. Zhang and S. Y. Fu, Dalton Trans., 2010, 39, 3426–3432 RSC.
- J. Shang, W. Hao, X. Lv, T. Wang, X. Wang, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, ACS Catal., 2014, 4, 954–961 CrossRef CAS.
- J. Ding, S. Sun, J. Bao, Z. Luo and C. Gao, Catal. Lett., 2009, 130, 147–153 CrossRef CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, Res. Chem. Intermed., 2005, 31, 513–519 CrossRef CAS PubMed.
- Y. Li, S. Dong, Y. Wang, J. Sun, Y. Li, Y. Pi, L. Hu and J. Sun, J. Mol. Catal. A: Chem., 2014, 387, 138–146 CrossRef CAS PubMed.
- S. Dong, Y. Cui, Y. Wang, Y. Li, L. Hu, J. Sun and J. Sun, Chem. Eng. J., 2014, 249, 102–110 CrossRef CAS PubMed.
- D. Channei, B. Inceesungvorn, N. Wetchakun and S. Phanichphant, J. Nanosci. Nanotechnol., 2014, 14, 7756–7762 CrossRef PubMed.
- L. Zhang, W. Wang, S. Sun, D. Jiang and E. Gao, CrystEngComm, 2013, 15, 10043–10048 RSC.
- X. Zhao, H. Liu, Y. Shen and J. Qu, Appl. Catal., B, 2011, 106, 63–68 CAS.
- S. Kant, S. Kalia and A. Kumar, J. Alloys Compd., 2013, 578, 249–256 CrossRef CAS PubMed.
- C. Singh, S. Bansal and S. Singhal, Phys. B, 2014, 444, 70–76 CrossRef CAS PubMed.
- W. Shi, J. Shi, S. Yu and P. Liu, Appl. Catal., B, 2013, 138, 184–190 CrossRef PubMed.
- W. Du, X. Wang, H. Li, D. Ma, S. Hou, J. Zhang, X. Qian and H. Pang, J. Am. Ceram. Soc., 2013, 96, 2979–2986 CrossRef CAS PubMed.
- J. M. Song, H. Q. Hu, X. Z. Wang, S. J. Zhao, Y. L. Shi and M. S. Ren, J. Inorg. Mater., 2013, 28, 1275–1280 CrossRef CAS.
- B. L. Fei, W. Li, J. H. Wang, Q. B. Liu, J. Y. Long, Y. G. Li, K. Z. Shao, Z. M. Su and W. Y. Sun, Dalton Trans., 2014, 43, 10005–10012 RSC.
- P. D. Kanhere, J. Zheng and Z. Chen, J. Phys. Chem. C, 2011, 115, 11846–11853 CAS.
- R. Xing, L. Wu, Z. Fei and P. Wu, J. Environ. Sci., 2013, 25, 1687–1695 CrossRef CAS.
- V. Štengl, S. Bakardjieva, N. Murafa, V. Houšková and K. Lang, Microporous Mesoporous Mater., 2008, 110, 370–378 CrossRef PubMed.
- N. Soltani, E. Saion, W. M. M. Yunus, M. Erfani, M. Navasery, G. Bahmanrokh and K. Rezaee, Appl. Surf. Sci., 2014, 290, 440–447 CrossRef CAS PubMed.
- C. Tan, G. Zhu, M. Hojamberdiev, K. S. Lokesh, X. Luo, L. Jin, J. Zhou and P. Liu, J. Hazard. Mater., 2014, 278, 572–583 CrossRef CAS PubMed.
- X. Zheng, S. Meng, J. Chen, J. Wang, J. Xian, Y. Shao, X. Fu and D. Li, J. Phys. Chem. C, 2013, 117, 21263–21273 CAS.
- C. Yu, L. Wei, J. Chen, Y. Xie, W. Zhou and Q. Fan, Ind. Eng. Chem. Res., 2014, 53, 5759–5766 CrossRef CAS.
- I. S. Cho, D. W. Kim, S. Lee, C. H. Kwak, S. T. Bae, J. H. Noh, S. H. Yoon, H. S. Jung, D. W. Kim and K. S. Hong, Adv. Funct. Mater., 2008, 18, 2154–2162 CrossRef CAS.
- K. Ding, B. Chen, Z. Fang and Y. Zhang, Theor. Chem. Acc., 2013, 132, 1352 CrossRef.
- X. Wang, G. Liu, Z.-G. Chen, F. Li, G. Q. Lu and H. M. Cheng, Chem. Lett., 2009, 38, 214–215 CrossRef.
- H. Guo, Y. Ke, D. Wang, K. Lin, R. Shen, J. Chen and W. Weng, J. Nanopart. Res., 2013, 15, 1475 CrossRef PubMed.
- S. Liang, R. Liang, L. Wen, R. Yuan, L. Wu and X. Fu, Appl. Catal., B, 2012, 125, 103–110 CrossRef CAS PubMed.
- W. J. Kim, D. Pradhan, B.-K. Min and Y. Sohn, Appl. Catal., B, 2014, 147, 711–725 CrossRef CAS PubMed.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 Search PubMed.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- Y. Li, W. Xie, X. Hu, G. Shen, X. Zhou, Y. Xiang, X. Zhao and P. Fang, Langmuir, 2010, 26, 591–597 CrossRef CAS PubMed.
- L. Luo, A. T. Cooper and M. Fan, J. Hazard. Mater., 2009, 161, 175–182 CrossRef CAS PubMed.
- Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wan and M. Fan, RSC Adv., 2014, 4, 13610–13619 RSC.
- S. T. Gadge and B. M. Bhanage, RSC Adv., 2014, 4, 10367–10389 RSC.
- J. An and Q. Zhou, J. Environ. Sci., 2012, 24, 827–833 CrossRef CAS.
- C. Zhao, M. Pelaez, X. Duan, H. Deng, K. O'Shea, D. Fatta-Kassinos and D. D. Dionysiou, Appl. Catal., B, 2013, 134–135, 83–92 CrossRef CAS PubMed.
- G. Li, Y. Zhang, L. Wu, F. Wu, R. Wang, D. Zhang, J. Zhu and H. Li, RSC Adv., 2012, 2, 4822–4828 RSC.
- S. Higashimoto, R. Shirai, Y. Osano, M. Azuma, H. Ohue, Y. Sakata and H. Kobayashi, J. Catal., 2014, 311, 137–143 CrossRef CAS PubMed.
- A. A. Ismail, L. Robben and D. W. Bahnemann, ChemPhysChem, 2011, 12, 982–991 CrossRef CAS PubMed.
- S. Higashimoto, Y. Hatada, R. Ishikawa, M. Azuma, Y. Sakata and H. Kobayashi, Curr. Org. Chem., 2013, 17, 2374–2381 CrossRef CAS.
- H. B. Y. Smida, M. Beicheickh and B. Jamoussi, J. Residuals Sci. Technol., 2013, 10, 47–54 CAS.
- R. M. Mohamed and E. Aazam, Desalin. Water Treat., 2013, 51, 6082–6090 CrossRef CAS.
- L. Wang, X. Li, W. Teng, Q. Zhao, Y. Shi, R. Yue and Y. Chen, J. Hazard. Mater., 2013, 244–245, 681–688 CrossRef CAS PubMed.
- J. Yang, J. Dai and J. Li, Environ. Sci. Pollut. Res., 2012, 20, 2435–2447 CrossRef PubMed.
- H. Chen, X. Zheng, Y. Chen and H. Mu, RSC Adv., 2013, 3, 9835–9842 RSC.
- K. S. Yao, D. Y. Wang, C. Y. Chang, K. W. Weng, L. Y. Yang, S. J. Lee, T. C. Cheng and C. C. Hwang, Surf. Coat. Technol., 2007, 202, 1329–1332 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, Q. Li, X. Zhang and J. K. Shang, ACS Appl. Mater. Interfaces, 2013, 5, 10953–10959 CAS.
- S. Dong, L. Hu, J. Feng, Y. Pi, Q. Li, Y. Li, M. Liu, J. Sun and J. Sun, RSC Adv., 2014, 4, 64994–65003 RSC.
| This journal is © The Royal Society of Chemistry 2015 |