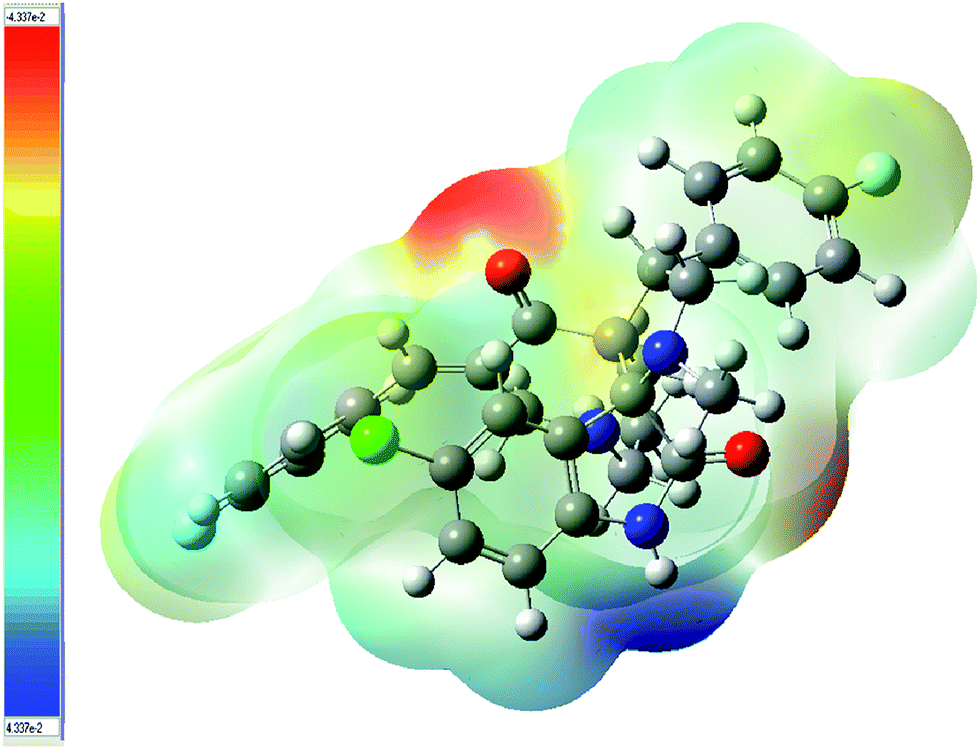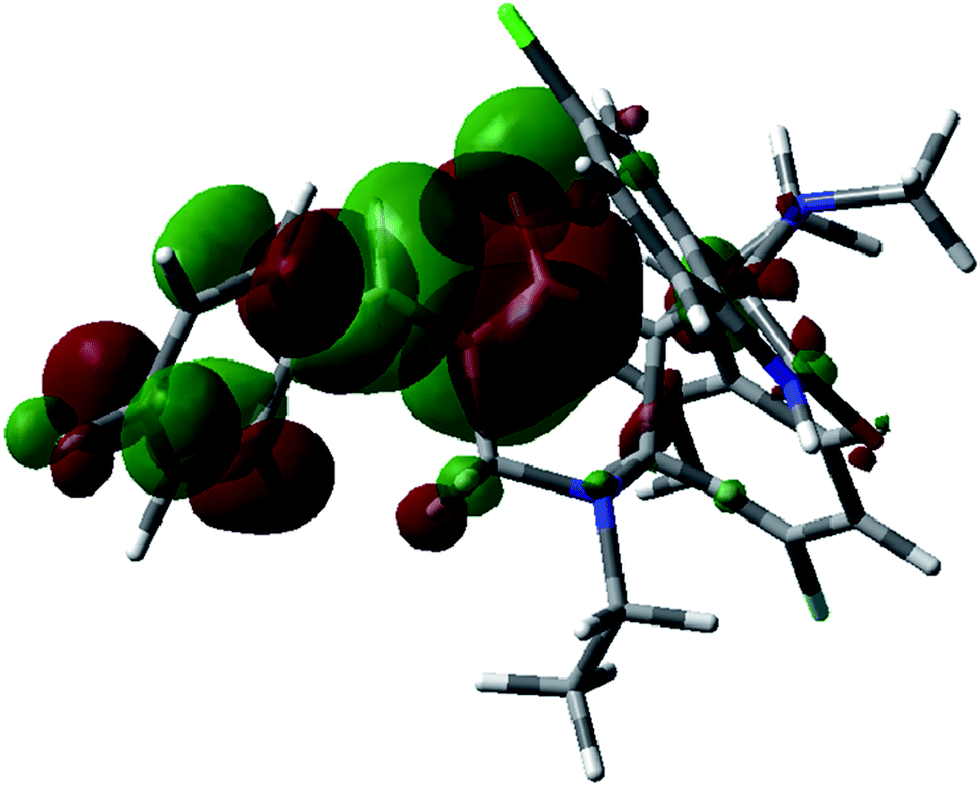DOI:
10.1039/C4RA13433H
(Communication)
RSC Adv., 2015,
5, 14780-14787
Regioselective synthesis and theoretical studies of an anti-neoplastic fluoro-substituted dispiro-oxindole†
Received
29th October 2014
, Accepted 13th January 2015
First published on 13th January 2015
Abstract
Single crystal X-ray studies of regioselectively synthesized fluoro-substituted dispiro-oxindole 9 revealed that the structure belongs to the monoclinic space group P21/n with four molecules in the unit cell. The structure was also investigated by AM1, PM3, and DFT studies. Compound 9 exhibits high potency against the HeLa cell line.
Introduction
Following cardiovascular disease, cancer is the second leading cause of death in both developed and developing countries.1 Cancer is a multigenic disease characterized by a diversity of genetic and epigenetic alterations and can be cured by preventing the rapid proliferation of cancer cells.2 Cancer accounted for 8.2 million deaths worldwide in 2012.3 The World Health Organization predicts a rise in deaths from cancer to 13.1 million in 2030.4 Cancer remains a challenging problem and treatment usually involves a combination of radiotherapy, surgery and chemotherapy. Chemotherapy is considered to be one of the effective approaches in suppressing tumor growth and eradication of tumors.5 There is still scope, however, for improvement of anticancer drugs, with respect to reduced toxicity, increased clinical effectiveness, broader spectrum of action, and elimination of side effects (e.g. nausea, hearing loss or vomiting).6
Indole alkaloids, found as natural products possess a broad spectrum of pharmacological action, in particularly anticancer activity. Several 2-oxindole derivatives are well known as antitumor agents due to their kinase inhibitory properties, especially tyrosine kinase inhibitory effects.7–9 Semaxanib (SU-5416) 1 and Sunitinib (SU-11248) 2 bearing a 2-oxiindole skeleton (Fig. 1) are the most promising tyrosine kinase inhibitors with antiangiogenic properties8 and are well-tolerated by human subjects.10,11 Mitraphylline 3, is a 2-oxindole analogue with a spiropyrrolidine-oxindole structural motif, isolated from Uncaria tomentosa, and possesses potent antitumor properties against human brain cancer cell lines, neuroblastoma SKN-BE(2) and malignant glioma GAMG.12 More examples of this family include spirotryprostatins A (4) and B (5), which were isolated from the fermentation broth of Aspergillus fumigatus, and completely inhibit G2/M progression cell division in mammalian tsFT210 cells13,14 (Fig. 1).
 |
| | Fig. 1 Antitumor active 2-oxindole containing compounds. | |
In the present work, the synthesis of fluoro-substituted dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine] is investigated. Single crystal X-ray studies identify the stereochemical structure of the prepared compound. X-ray diffraction of single crystals has the capacity to distinguish and identify the enantiomers/diasteromers of molecules.15 The present work also includes geometry optimization of the synthesized compound utilizing different computational chemistry techniques (AM1, PM3 and DFT). This allows comparison of the experimental and theoretical data. Studies of molecular electrostatic potential (MEP) and frontier molecular orbitals (FMOs) are also considered.
The antitumor properties of the synthesized dispiroindole analogue were screened against human tumor cell lines, including HeLa (cervical), and HepG2 (liver) cancers and the rationale for this is as follows. Hepatocellular carcinoma causes 662![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths worldwide per year. Overall, liver cancer is the third most common cause of cancer death after lung and stomach cancer.3,16 To date, surgical resection is the most effective treatment option, but is available only to a small number of patients, and the rate of recurrence is high. Chemotherapy is also used for liver cancer patients who are not suitable for surgery, or is applied as treatment in addition to surgery. However, severe toxic side effects, low tumor-selectivity, and the highly metastatic and chemo-resistant nature of liver cancer greatly hamper the effectiveness of chemotherapy.17 Hepatocellular carcinoma usually occurs in patients with chronic liver disease. Infection with hepatitis B or C virus (HBV, HCV) is the leading cause of this type of cancer, with each virus infection increasing the risk of cancer more than 10-fold.18 HCV is considered a national problem in many countries and is responsible for the elevated number of hepatocellular carcinoma observed in recent years. The other malignant disease targeted by the synthesized alkaloid analogue is cervical carcinoma, which is the third most common type of cancer in women and is almost always caused by human papillomavirus (HPV) infection.19,20 The synthesized compound is characterized by the presence of a fluorine substituent due to the unique properties of fluorine-containing compounds in terms of high thermal stability and lipophilicity.21,22
000 deaths worldwide per year. Overall, liver cancer is the third most common cause of cancer death after lung and stomach cancer.3,16 To date, surgical resection is the most effective treatment option, but is available only to a small number of patients, and the rate of recurrence is high. Chemotherapy is also used for liver cancer patients who are not suitable for surgery, or is applied as treatment in addition to surgery. However, severe toxic side effects, low tumor-selectivity, and the highly metastatic and chemo-resistant nature of liver cancer greatly hamper the effectiveness of chemotherapy.17 Hepatocellular carcinoma usually occurs in patients with chronic liver disease. Infection with hepatitis B or C virus (HBV, HCV) is the leading cause of this type of cancer, with each virus infection increasing the risk of cancer more than 10-fold.18 HCV is considered a national problem in many countries and is responsible for the elevated number of hepatocellular carcinoma observed in recent years. The other malignant disease targeted by the synthesized alkaloid analogue is cervical carcinoma, which is the third most common type of cancer in women and is almost always caused by human papillomavirus (HPV) infection.19,20 The synthesized compound is characterized by the presence of a fluorine substituent due to the unique properties of fluorine-containing compounds in terms of high thermal stability and lipophilicity.21,22
Results and discussion
Chemistry
1,3-Dipolar cycloaddition reaction of non-stabilized azomethine ylide, (generated in situ via decarboxylative condensation of 5-chloroisatin 6 with sarcosine 7) and 3E,5E-3,5-bis[(4-fluorophenyl)methylidene]-1-ethyl-4-piperidone 8 in refluxing ethanol proceeds regioselectively by affording a single product as monitored by TLC. The structure of the isolated colorless product was assigned as 5-chloro-1′′-ethyl-1′-methyl-4′-(4-fluorophenyl)-5′′-[(4-fluorophenyl)methylidene]-dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine]-2(1H),4′′-dione 9 based on spectroscopic (IR, 1H-, 13C-NMR, 1H, 1H-COSY, HSQC, HRMS) evidence. The reaction commences by nucleophilic attack of the amino group of sarcosine 7 on the 3-carbonyl function of 5-chloroisatin 6, followed by dehydration to form a spiro-oxazalidinone system. This expels carbon dioxide to generate a reactive, non-stabilized azomethine ylide, that undergoes in situ 1,3-dipolar cycloaddition with the exocyclic olefinic linkage of piperidone 8 eventually affording 9 (Scheme 1).
 |
| | Scheme 1 Synthetic route towards dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine] 9. | |
The IR spectrum of 9 (Fig. S1 of ESI†) shows the indolyl amidic NH stretching vibration band at ν = 3157 cm−1 in addition to strong stretching vibration bands at ν = 1701, 1678 cm−1 which are assigned to the ketonic and amidic carbonyl functions. The 1H-NMR spectrum of 9 (Fig. S2 of ESI†) reveals the methylene protons H2C-5′, H2C-2′′ and H2C-6′′ as diastereotopic two spin systems, and the methine HC-4′ appears as a double doublet signal at δ = 4.62, due to coupling with the diastereotopic pyrrolidinyl H2C-5′. The methylene protons of the ethyl function attached to the piperidinyl N-1′′ appear as non-equivalent protons (diastereotopic protons, δ = 2.11, 2.24) exhibiting coupling with each other and in turn with the vicinal methyl protons. The 1H, 1H-COSY spectrum of compound 9 (Fig. S3 of ESI†) supports these interpretations. The 13C-NMR spectrum of 9 (Fig. S4 of ESI†) exhibits the methylene carbons H2C-6′′, H2C-2′′ and H2C-5′ at δ = 53.2, 56.1, and 56.7, respectively. The methine HC-4′ appears at δ = 44.2 and the ethyl carbons attached to the piperidinyl N-1′′ appear at δ = 10.9, 51.1. Meanwhile, the methyl carbon attached to the pyrrolidinyl N-1′ appears at δ = 34.1. The spiro-carbons C-3′ (C-3′′) and C-3 (C-2′) appear at δ = 64.5, and 75.3, respectively. The carbonyl carbons C-2 and C-4′′ are at δ = 176.1, and 198.0, respectively (c.f. experimental section). The 1H, 13C-Heteronuclear Single Quantum Coherence (HSQC) spectrum of compound 9 supports these assignments (Fig. S5 and S6 of ESI†).
Single crystal X-ray
Compound 9 crystallizes in the monoclinic space group P21/n with four molecules in the unit cell, which contains both enantiomers of compound 9, (see Fig. 2 for an ORTEP view of 9). Two spiro carbon/linkages are exhibited at C26, where the piperidine and pyrrolidine rings are connected, and at C27, where the pyrrolidine and indole rings are attached. Furthermore, the pyrrolidine-bound aryl ring is attached at C21. In general, the bond lengths and angles (Tables S1 and S2 of ESI†) are in good agreement with previously determined structures having similar molecular architecture.23–27 The weighted average ring bond distances C12 → C13 → C33 → C24 → C34 → C18, C14 → C15 → C25 → C22 → C17 → C16 and C30 → C32 → C35 → C31 → C36 → C38 are 1.386(5), 1.388(2) and 1.389(4) Å, respectively.
 |
| | Fig. 2 An ORTEP view of compound 9 showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii. | |
The indolyl group as well as the 4-fluorophenyl ring (C12 → C13 → C33 → C24 → C34 → C18 and C14 → C15 → C25 → C22 → C17 → C16) have planar configurations with maximum deviation of 0.046(2) Å for atom N5, 0.031(2) Å for atom C12 and 0.027(2) Å for atom C17. The (4-fluorophenyl)methylidene moiety is connected to the piperidine ring at position C9. The double bond of C9–C11 has the E-configuration. The sum of the angles around the piperidine-N7 and the pyrrolidine-N6 atoms are approximately 330° and 335°, respectively, confirming their sp3 character. The piperidine ring adopts a half-chair conformation where the C10 atom lies 0.702(3) Å out of the plane of the remaining five atoms. The C27 and C21 atoms occupy axial and equatorial positions with respect to the piperidine ring. Meanwhile, the (4-fluorophenyl)methylidene moiety occupies an equatorial position, and the N-bound ethyl substituent is equatorial. The pyrrolidine ring has an envelope conformation with the flap atom being N6 which lies 0.582(3) Å out of the plane of the remaining four atoms.
Centrosymmetric eight-membered {⋯HNCO}2 amide dimer formation is the most significant feature of the crystal packing of 9 (Fig. 3). In this crystal structure, molecules are linked together by intermolecular C–H⋯O, C–H⋯F and N–H⋯O hydrogen-bonding (Table 1). The crystal structure is further stabilized by an intermolecular C–H⋯π (π-ring) stacking interaction (C28–H28B⋯Cg1, C29–H29A⋯Cg2 and C37–H37A⋯Cg3), where the Cg1 ring refers to C30/C32/C35/C31/C36/C38, the Cg2 ring to C12/C13/C33/C24/C34/C18 and the Cg3 ring to N5/C16/C17/C27/C20, Fig. 3.
 |
| | Fig. 3 Crystal packing in the unit cell of compound 9 showing a view in projection down the a axis of the unit cell with the different hydrogen-bond interactions shown as colored lines. | |
Table 1 Hydrogen-bond geometry (Å, °) for compound 9a
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
D–H⋯A |
| Symmetry codes: (i) 1 − x, −y, 1 − z; (ii) x, y, 1 + z; (iii) 3/2 − x, 1/2 + y, 1/2 − z; (iv) −1/2 + x, −1/2 − y, −1/2 + z, (v) −1/2 + x, −1/2 − y, 1/2 + z, (vi) x, y, z. |
| N5–H5⋯O1i |
0.87(2) |
1.94(2) |
2.812(2) |
172.6(19) |
| C31–H31A⋯F2ii |
0.95 |
2.49 |
3.095(2) |
121 |
| C33–H33A⋯F4iii |
0.95 |
2.52 |
3.299(2) |
139 |
| C34–H34A⋯O3iv |
0.95 |
2.26 |
3.192(2) |
167 |
| C28–H28B⋯Cg1iv |
0.95 |
2.78 |
3.713(2) |
158 |
| C29–H29A⋯Cg2v |
0.95 |
2.88 |
3.610(2) |
132 |
| C37–H37A⋯Cg3vi |
0.95 |
2.73 |
2.879(2) |
89 |
AM1, PM3 and DFT studies
Theoretical calculations using both AM1 and PM3 methods were undertaken for compound 9 to compare the geometrical parameters obtained from the single crystal X-ray analysis with the calculated data. The geometry of 9 was optimized by the molecular mechanics force field (MM+), followed by either semi-empirical AM1 or PM3 methods implemented in the HyperChem 8.0 package. The structure was fully optimized without fixing any parameters, thus bringing all geometric variables to their equilibrium values. The energy minimization protocol employed the Polake–Ribiere conjugated gradient algorithm. Convergence to a local minimum was achieved when the energy gradient was ≤0.01 kcal mol−1. The RHF method was used in the spin pairing for the two semi-empirical tools27–30 (Fig. S7 and S8 of ESI† exhibit the optimized structure of 9 by AM1 and PM3, respectively). Additionally, the molecular structure of 9 in the ground state (in vacuo) was optimized using density functional theory (DFT) as originally proposed by Hohenberg and Kohn, starting from the experimental structure.31–33 DFT calculations with a hybrid functional B3LYP [Becke's three parameter hybrid functional using the Lee–Yang–Parr (LYP) correlation functional]34,35 at 3-21G* basis set utilizing the Gaussian 03 package.27b The geometries were optimized by minimizing energies with respect to all the geometrical parameters without imposing any molecular symmetry constraints (Fig. S9 of ESI†). In DFT the energy of the molecule is determined from the electron density instead of a wave function and has emerged as an important quantum chemical tool for studying various chemical problems.36,37 Tables S1 and S2 of the ESI† list the intramolecular experimentally (single crystal X-ray) and theoretically calculated parameters (bond lengths and angles) utilizing the AM1, PM3, and DFT methods for compound 9.
Comparing the geometrical parameters delivered from the optimized structure by the AM1, PM3, and DFT methods with the experimental X-ray data, most of the bond lengths and angles of the optimized structures showed a good fit with experiment. Differences can be attributed to the fact that the experimental results were obtained for the solid phase whereas the theoretical calculations were for the in vacuo (gas phase) structure. In the solid state, the existence of a crystal field along with intermolecular interactions connect the molecules in the lattice which affects the bond parameters.38 The biggest differences between the experimental and calculated bond lengths using the AM1, PM3 and DFT methods are 0.053, 0.092, and 0.038 Å, respectively. Moreover, the root mean square errors (RMSE) are 0.0191, 0.0223, and 0.0132 Å for AM1, PM3 and DFT techniques, respectively. These statistical observations indicate that the bond lengths obtained by the three optimization methods, especially the DFT method, correlate strongly with the experimentally observed values.
Regarding the bond angles, the biggest differences between the experimental and calculated values are 5.7, 5.8, and 5.68° and the root mean square errors (RMSE) are 1.924, 1.818, and 1.379° for AM1, PM3, and DFT methods, respectively. Global comparisons were performed by superimposing the molecular skeletons obtained from X-ray diffraction with the corresponding ones of the AM1, PM3, and DFT methods (Fig. 4). It can be seen that most of the functions align well with each other. However, the ethyl group attached to the piperidinyl nitrogen in the theoretically optimized structures (AM1, PM3 and DFT) is aligned in a different direction to that observed by X-ray. This is obvious from the torsion angle values of C23–N7–C28–C29 (single X-ray data = 164.53°, however, for AM1, PM3, and DFT the values are 68.43, 60.06, and 66.60°, respectively). A slight difference is also noticed for the aryl group attached to the exocylic olefinic linkage. The torsion angle of C18–C12–C11–C9 for the single X-ray data = 29.28°; however, AM1, PM3, and DFD give −44.73, −47.79, and −13.46°, respectively. These observations can be attributed to the effect of lattice form affecting the X-ray structural behavior. Whereas in the theoretical studies by AM1, PM3, or DFT this effect is completely neglected.
 |
| | Fig. 4 Overlay diagram of compound 9, drawn so that the central pyrrolidine rings are overlapped, red (single X-ray structure), green (AM1), blue (PM3), and yellow (DFT). | |
Molecular electrostatic potential
Molecular electrostatic potential (MEP) is a very useful property for analyzing and predicting molecular behavior. It is a good guide in assessing reactivity towards positively or negatively charged reactants and hydrogen bonding interactions. MEP surfaces depict the size, shape and variation of electron density and correlate with dipole moment, electronegativity, partial charges and the sites of chemical reactivity within the molecule. The electrostatic potential V(r) is also well suited for analyzing processes based on the recognition of one molecule by another, as in drug–receptor, and enzyme–substrate interactions.39 This surface represents the distance from a molecule at which a positive test charge experiences a certain amount of attraction or repulsion and they highlight the bonding possibilities during complex formation. Molecular electrostatic potential V(r) is defined by eqn (1).| |
 | (1) |
where ZA is the charge of nucleus A, located RA, ρ(r′) is the electronic density function of the molecule and r′ is a dummy integration variable.39 MEP was calculated for compound 9 using B3LYP/6-31G(d,p). The MEP diagram of 9 is shown in Fig. 5. The resulting overall MEP of 9 leaves no doubt that the intense (negative) red color around the carbonyl group of the piperidone ring is the most electrophilic site. However, the (positive) blue region, which is related to the most nucleophilic reactive center, is mainly located around the indolyl nitrogen.
 |
| | Fig. 5 Molecular electrostatic potential map (MEP) of compound 9 calculated at the B3LYP/6-31G(d,p) level (in a.u.). | |
Frontier molecular orbitals
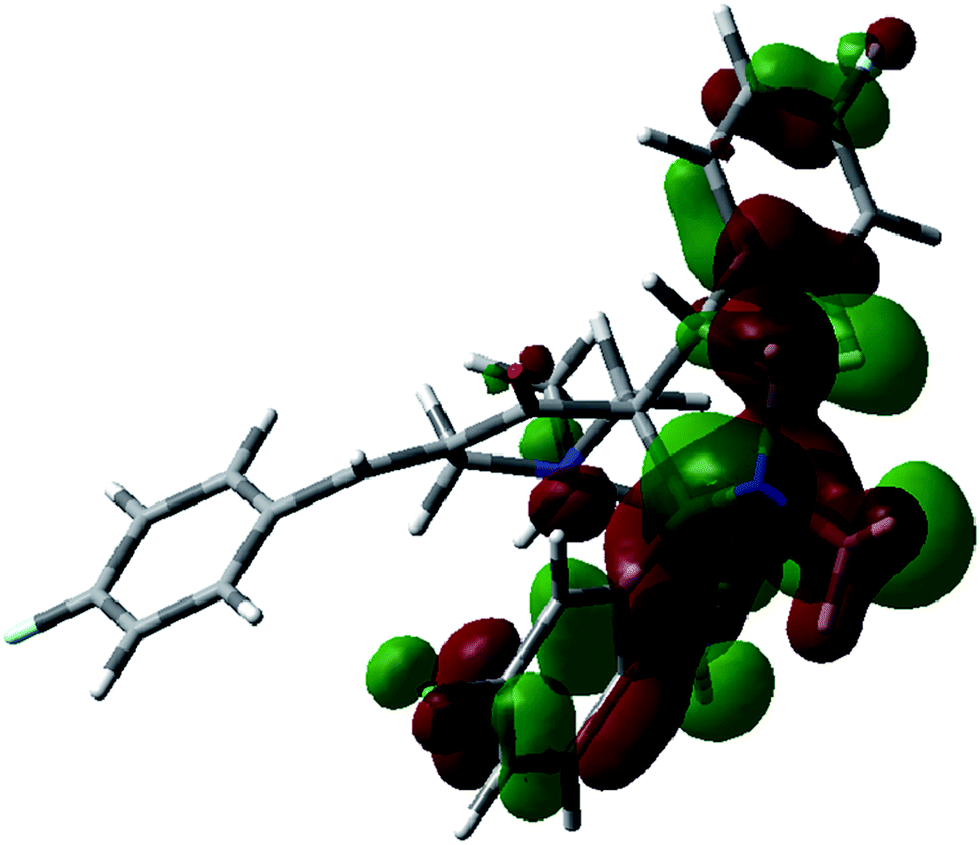
Frontier molecular orbitals (FMOs) play an important role in electric and optical properties, in UV-visible spectra and chemical reactions. The HOMO and LUMO are also important in determining such properties as molecular reactivity and the ability of a molecule to absorb light. The LUMO (lowest unoccupied molecular orbital) is the molecular orbital of lowest energy that is not occupied by electrons and represents the ability to accept an electron. The HOMO (highest occupied molecular orbital) is the molecular orbital of highest energy that is occupied by electrons and represents the ability to donate an electron.39 FMO calculations for compound 9 were undertaken utilizing the Gaussian 03 program package at the B3LYP/6-31G(d,p) level of theory. The results are shown in Fig. 6, and 7 together with the energy levels. The indole and pyrrolidine rings mainly contribute to the HOMO of 9, while the (4-fluorophenyl)methylidene residue together with the piperidine ring contribute to the LUMO. The energy difference between the HOMO (−0.207 eV) and LUMO (−0.075 eV) in compound 9 is 0.132 eV. The energy gap between HOMO and LUMO indicates high kinetic stability, and high excitation energies for many of the excited states.
 |
| | Fig. 6 Molecular orbital surfaces of energy level (−0.207 eV) for the HOMO of compound 9 computed by the B3LYP/6-31G(d,p) method. | |
 |
| | Fig. 7 Molecular orbital surfaces of energy level (−0.075 eV) for the LUMO of compound 9 computed by the B3LYP/6-31G(d,p) method. | |
Antitumor properties
The antitumor properties of compound 9 were screened against HepG2 (liver) and HeLa (cervical) human tumor cell lines utilizing the in vitro Sulfo-Rhodamine-B (SRB) standard method.28,40–45 From the results obtained (Table 2, Fig. S10 and S11 of ESI†), it is apparent that compound 9 shows moderate antitumor properties against HepG2 (hepatocellular carcinoma) with IC50 (concentration required to produce 50% inhibition of cell growth) = 12.50 μM, compared with the IC50 of standard references used, doxorubicin hydrochloride = 8.05, and cisplatin = 11.89 μM, respectively. However, compound 9 exhibits higher potency against HeLa (cervical carcinoma) cell line (IC50 = 5.75 μM) than doxorubicin hydrochloride and cisplatin (IC50 = 7.22, 7.71 μM, respectively). Cisplatin is the drug of choice either alone or in combination with topotecan46 for cervical chemotherapy and cisplatin in combination with 5-fluorouracil has also been reported.47,48 However, severe side-effects such as bone-marrow depression, neutropenia, thrombocytopenia and anaemia due to haematological toxicity along with nephrotoxicity and neurotoxicity49 and acquired chemoresistance50 throughout the course of treatment, have limited the usage of cisplatin.
Table 2 Antitumor properties of compound 9
| Entry |
Compound |
IC50a, μg ml−1 (μM) |
| HepG2 (liver) |
HeLa (cervical) |
| IC50 = concentration required to produce 50% inhibition of cell growth compared to control experimental. |
| 1 |
9 |
6.85 (12.50) |
3.15 (5.75) |
| 2 |
Doxorubicin hydrochloride |
4.67 (8.05) |
4.19 (7.22) |
| 3 |
Cisplatin |
3.58 (11.89) |
4.19 (7.71) |
Conclusions
In conclusion, the 1,3-dipolar cycloaddition reaction of non-stabilized azomethine ylide, (generated in situ via decarboxylative condensation of 5-chloroisatin 6 with sarcosine 7) and 3E,5E-3,5-bis[(4-fluorophenyl)methylidene]-1-ethyl-4-piperidone 8 proceeds regioselectively to afford the corresponding 5-chloro-1′′-ethyl-1′-methyl-4′-(4-fluorophenyl)-5′′-[(4-fluorophenyl)methylidene]-dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine]-2(1H),4′′-dione 9 as the sole product in good yield (73% yield). Single crystal X-ray studies of 9 revealed its stereochemical structure and purity. Theoretical calculations using AM1, PM3, and DFT (B3LYP/3-21G*) methods of compound 9 show most of the geometrical parameters, including bond lengths and angles of the optimized structures, fit well with experiment. The notable differences due to torsion angle of the theoretically optimized structures (AM1, PM3 and DFT) and experimental observed X-ray for the ethyl group attached to the piperidinyl nitrogen and aryl group attached to the exocylic olefinic linkage are attributed to the fact that the theoretical calculations were obtained in vacuo (gas phase) studies whilst the experimental results were obtained from the solid phase studies. The MEP of 9 calculated utilizing DFT-B3LYP/6-31G(d,p) showed that the most electrophilic site is the carbonyl group of the piperidone ring and the most nucleophilic reactive center is located around the indolyl nitrogen. Additionally, FMOs of 9 were determined revealing the energy difference between the HOMO and LUMO to be 0.132 eV. Compound 9 exhibits higher potency against the HeLa (cervical carcinoma) cell line than those of doxorubicin hydrochloride and cisplatin. However, the antitumor activity against HepG2 (hepatocellular carcinoma) was less than that of the standards.
Experimental section
General
Melting points were determined on a capillary point apparatus equipped with a digital thermometer. IR spectra (KBr) were recorded on a Shimadzu FT-IR 8400S spectrophotometer. NMR spectra were recorded in DMSO-d6 on a Mercury NMR spectrometer operating at 300 MHz for 1H (with TMS as an internal standard) and 75 MHz for 13C. High-resolution mass spectra (HRMS) were recorded on an Agilent Technologies 6210 Time of Flight LC/MS instrument operating in the ESI mode. The starting compound 8 was prepared according to the previously reported procedure.51
Synthesis of 5-chloro-1′′-ethyl-1′-methyl-4′-(4-fluorophenyl)-5′′-[(4-fluorophenyl)methylidene]-dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine]-2(1H),4′′-dione (9). Equimolar quantities of 3E,5E-3,5-bis[(4-fluorophenyl)methylidene]-1-ethyl-4-piperidone 8 (2 mmol), 5-chloroisatin 6 and sarcosine 7 were heated under reflux for 10 h in ethanol (25 ml). The separated solid after storing the reaction mixture overnight at room temperature was collected and crystallized from methanol affording the corresponding 5-chloro-1′′-ethyl-1′-methyl-4′-(4-fluorophenyl)-5′′-[(4-fluorophenyl)methylidene]-dispiro[3H-indole-3,2′-pyrrolidine-3′,3′′-piperidine]-2(1H),4′′-dione (9) as colorless microcrystals. Mp 210–212 °C, yield (0.80 g) 73%. IR: νmax cm−1 3157, 1701, 1678, 1599, 1585. 1H-NMR: δ 0.76 (t, J = 7.1 Hz, 3H, piperidinyl NCH2CH3), 1.64 (d, J = 12.5 Hz, 1H, upfield H of piperidinyl H2C-2′′), 1.98 (s, 3H, pyrrolidinyl NCH3), 2.11 (dd, J = 12.1, 6.9 Hz, 1H, upfield H of piperidinyl NCH2CH3), 2.24 (dd, J = 12.1, 7.3 Hz, 1H, downfield H of piperidinyl NCH2CH3), 2.95 (d, J = 13.9 Hz, 1H, upfield H of piperidinyl H2C-6′′), 3.10–3.34 (m, 3H, downfield H of piperidinyl H2C-2′′ + upfield H of pyrrolidinyl H2C-5′ + downfield H of piperidinyl H2C-6′′), 3.74 (t, J = 9.7 Hz, 1H, downfield H of pyrrolidinyl H2C-5′), 4.62 (dd, J = 10.7, 7.5 Hz, 1H, pyrrolidinyl HC-4′), 6.61 (d, J = 8.3 Hz, 1H, arom. H), 6.78 (d, J = 2.1 Hz, 1H, arom. H), 7.08 (dd, J = 8.3, 2.3 Hz, 1H, arom. H), 7.11–7.28 (m, 7H, arom. H), 7.35 (dd, J = 8.6, 5.6 Hz, 2H, arom. H), 10.55 (s, 1H, NH). 13C-NMR: δ 10.9 (piperidinyl NCH2CH3), 34.1 (pyrrolidinyl NCH3), 44.2 (pyrrolidinyl HC-4′), 51.1 (piperidinyl NCH2CH3), 53.2 (piperidinyl H2C-6′′), 56.1 (piperidinyl H2C-2′′), 56.7 (pyrrolidinyl H2C-5′), 64.5 [spiro C-3′ (C-3′′)], 75.3 [spiro C-3 (C-2′)], 110.1, 114.9, 115.1, 115.5, 115.8, 124.8, 126.7, 128.4, 128.9, 130.8, 130.9, 131.0, 132.3, 132.4, 132.92, 132.94, 134.3, 134.31, 135.9, 142.4, 159.5, 160.5, 162.7, 163.8 (arom. C + olefinic C), 176.1 [indolyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (C-2)], 198.0 [piperidinyl CO (C-4′′)]. HRMS (ESI): m/z [M + H]+ calcd for C31H29ClF2N3O2: 548.1911; found: 548.1902.
O (C-2)], 198.0 [piperidinyl CO (C-4′′)]. HRMS (ESI): m/z [M + H]+ calcd for C31H29ClF2N3O2: 548.1911; found: 548.1902.
Single crystal X-ray
The X-ray single crystal diffraction data were collected at 120 K on an Agilent SuperNova instrument with focussed microsource Cu Kα radiation (λ = 1.5418 Å) and ATLAS CCD area detector. Details of the data collection conditions and the parameters of the refinement are given in Table 3. The structure was solved using direct methods with SHELXS52 and refined on F2 using all data by full-matrix least square procedures with SHELXL-97.52 Multiscan absorption corrections were done using SCALE3 ABSPACK. The non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were included in calculated positions with isotropic displacement parameters 1.2 times the isotropic equivalent of their carrier atoms (Table 3).
Table 3 Crystal data and structure refinement parameters for compound 9
| Chemical formula |
C31H28ClF2N3O2 |
| Mr |
548.01 |
| Crystal system, space group |
Monoclinic, P21/n |
| Temperature (K) |
120 |
| a, b, c (Å) |
11.97300 (13), 16.1952 (3), 14.09653 (16) |
| β (°) |
93.4721 (10) |
| V (Å3) |
2728.37 (6) |
| Z |
4 |
| Radiation type |
Cu Kα |
| μ (mm−1) |
1.64 |
| Crystal size (mm) |
0.30 × 0.24 × 0.06 |
| Diffractometer |
SuperNova (Cu) X-ray diffractometer |
| Tmin, Tmax |
0.721, 0.922 |
| No. of measured, independent and observed [I > 2σ(I)] reflections |
38016, 4913, 4248 |
| Rint |
0.048 |
| (sinθ/λ)max (Å−1) |
0.599 |
| R[F2 > 2σ(F2)], wR(F2), S |
0.036, 0.096, 1.05 |
| No. of reflections |
4913 |
| No. of parameters |
356 |
| Δρmax, Δρmin (e Å−3) |
0.42, −0.23 |
Antitumor activity screening
Antitumor properties of compound 9 were screened by the National Cancer Institute, Cairo University, Egypt, using previously reported in vitro Sulfo-Rhodamine-B (SRB) standard technique for HepG2 (liver), HeLa (cervical) human tumor cell lines.28,40–45 Cells were seeded in 96-well microtiter plates at a concentration of 5 × 104 to 105 cell per well in a fresh medium and left for 24 h before treatment with the test compound to allow attachment of cells to the wall of the plate. The test compound was dissolved in dimethylsulfoxide (DMSO) and diluted 1000-fold in the assay. Different concentrations of the compound under test (0, 2.5, 6.25, 12.5, and 25 μg ml−1) were added to the cell monolayer. Triplicate wells were prepared for each individual dose. The monolayer cells were incubated with the test compound for 48 h at 37 °C, in an atmosphere of 5% CO2. After 48 h, the cells were fixed, washed and stained with Sulfo-Rhodamine-B (SRB) stain. Excess stain was washed with acetic acid. The attached stain was recovered with Tris–EDTA buffer. Cell survival and drug activity were determined by measuring the color intensity spectrophotometrically at 564 nm using an ELISA microplate reader (Meter tech. Σ 960, USA). Data are collected as mean values for experiments that were performed in three replicates for each individual dose which were measured by SRB assay. Control experiments did not exhibit significant change compared to the DMSO vehicle. Doxorubicin hydrochloride and cisplatin were used as standard references during the present in vitro bioactivity screening assay. The percentage of cell survival was calculated as follows:
| Surviving fraction = Optical density (O.D.) of treated cells/O.D. of control cells |
The IC50 (concentration required to produce 50% inhibition of cell growth compared to control experiment) was determined using Graph-Pad PRISM version-5 software. Statistical calculations for determination of the mean and standard error values were determined using SPSS 16 software. The observed antitumor properties are presented in Table 2 (Fig. S10 and S11 of ESI†).
Notes and references
- J. Y. Zhang, Nat. Rev. Drug Discovery, 2002, 1, 101–102 CrossRef CAS.
- T. Helleday, E. Petermann, C. Lundin, B. Hodgson and R. A. Sharma, Nat. Rev. Cancer, 2008, 8, 193–204 CrossRef CAS PubMed.
- H. K. Maurya, S. K. Gautam, R. Pratap, V. K. Tandon, A. Kumar, B. Kumar, S. Saxena, D. Tripathi, M. Rajwanshi, M. Das and V. J. Ram, Eur. J. Med. Chem., 2014, 81, 367–377 CrossRef CAS PubMed.
- http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
- C. W. Mai, M. Yaeghoobi, N. Abd-Rahman, Y. B. Kang and M. R. Pichika, Eur. J. Med. Chem., 2014, 77, 378–387 CrossRef CAS PubMed.
- E.-J. Gao, H. Fu, M.-C. Zhu, C. Ma, S.-K. Liang, J. Zhang, L.-F. Li, L. Wang, Y.-Y. Li and W. Jiao, Eur. J. Med. Chem., 2014, 82, 172–180 CrossRef CAS PubMed.
- R. D. Connell, Expert Opin. Ther. Pat., 2003, 13, 737–749 CrossRef CAS.
- M. A. Jianguo, L. I. Shaolan, K. Reed, P. Guo and J. M. Gallo, J. Pharmcol. Exp. Ther., 2003, 305, 833–839 CrossRef PubMed.
- P. Marzola, A. Degrassi, L. Calderan, P. Farace, C. Crescimanno, E. Nicolato, A. Giusti, E. Pesenti, A. Terron, A. Sbarbati, T. Abrams, L. Murray and F. Osculati, Clin. Cancer Res., 2004, 10, 739–750 CrossRef CAS.
- S. N. Pandeya, S. Smitha, M. Jyoti and S. K. Sridhar, Acta Pharm., 2005, 55, 27–46 CAS.
- K. L. Vine, L. Matesic, J. M. Locke, M. Ranson and D. Skropeta, Anti-Cancer Agents Med. Chem., 2009, 9, 397–414 CrossRef CAS.
- E. Garcia Prado, M. D. Garcia Gimenez, R. De la Puerta Vázquez, J. L. Espartero Sánchez and M. T. Sáenz Rodriguez, Phytomedicine, 2007, 14, 280–284 CrossRef CAS PubMed.
- C. B. Cui, H. Kakeya and H. Osada, Tetrahedron, 1996, 52, 12651–12666 CrossRef CAS.
- C. B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 832–835 CrossRef CAS.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- “Cancer” World Health Organization, February 2010, Retrieved 2011-01-05.
- M. Schwartz, S. Roayaie and M. Konstadoulakis, Nat. Clin. Pract. Oncol., 2007, 4, 424–432 CrossRef CAS PubMed.
- F. Donato, P. Boffetta and M. Puoti, Int. J. Cancer, 1998, 75, 347–354 CrossRef CAS.
- http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001895/.
- http://www.cancer.gov/cancertopics/types/cervical.
- N. Mishriky, F. M. Asaad, Y. A. Ibrahim and A. S. Girgis, Recl. Trav. Chim. Pays-Bas, 1994, 113, 35–39 CrossRef CAS.
- T. Haga, K. Fujikawa, T. Koyanagi, T. Nakajima and K. Hayashi, Heterocycles, 1984, 22, 117–124 CrossRef CAS.
- I. S. Ahmed Farag, A. S. Girgis, A. A. Ramadan, A. M. Moustafaa and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o22–o23 Search PubMed.
- I. S. Ahmed Farag, A. S. Girgis, A. A. Ramadan, A. M. Moustafaa and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o43–o44 Search PubMed.
- I. S. Ahmed Farag, A. S. Girgis, A. A. Ramadan, A. M. Moustafaa and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o70–o71 Search PubMed.
- I. S. Ahmed Farag, A. S. Girgis, A. A. Ramadan, A. M. Moustafa and A. F. Mabied, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o379–o380 Search PubMed.
-
(a) I. S. Ahmed Farag, A. S. Girgis, A. M. Moustafa, B. E. M. El-Gendy, A. F. Mabied and E. M. Shalaby, J. Mol. Struct., 2014, 1075, 327–334 CrossRef PubMed;
(b) E. M. Shalaby, A. S. Girgis, M. A. Ibrahim, N. S. M. Ismail, A. F. Mabied and I. S. Ahmed Farag, RSC Adv. Search PubMed , submitted.
- A. S. Girgis, J. Stawinski, N. S. M. Ismail and H. Farag, Eur. J. Med. Chem., 2012, 47, 312–322 CrossRef CAS PubMed.
- A. S. Girgis, N. S. M. Ismail and H. Farag, Eur. J. Med. Chem., 2011, 46, 2397–2407 CrossRef CAS PubMed.
- A. S. Girgis, H. Farag, N. S. M. Ismail and R. F. George, Eur. J. Med. Chem., 2011, 46, 4964–4969 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B846–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- P. Rawat and R. N. Singh, J. Mol. Struct., 2014, 1074, 201–212 CrossRef CAS PubMed.
- M. Jablonski and M. Palusiak, J. Phys. Chem. A, 2010, 114, 2240–2244 CrossRef CAS PubMed.
- E. İnkaya, M. Dinçer, Ö. Ekici and A. Cukurovali, Spectrochim. Acta, Part A, 2013, 101, 218–227 CrossRef PubMed.
- L. A. Taib, H. M. Faidallah, Z. S. Şahin, A. M. Asiri, O. Şahin and M. N. Arshad, J. Mol. Struct., 2014, 1076, 272–279 CrossRef CAS PubMed.
- R. F. George, N. S. M. Ismail, J. Stawinski and A. S. Girgis, Eur. J. Med. Chem., 2013, 68, 339–351 CrossRef CAS PubMed.
- A. M. Moustafa, A. S. Girgis, S. M. Shalaby and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o2197–o2198 CAS.
- A. R. Katritzky, A. S. Girgis, S. Slavov, S. R. Tala and I. Stoyanova-Slavova, Eur. J. Med. Chem., 2010, 45, 5183–5199 CrossRef CAS PubMed.
- A. S. Girgis, Eur. J. Med. Chem., 2009, 44, 1257–1264 CrossRef CAS PubMed.
- A. S. Girgis, Eur. J. Med. Chem., 2009, 44, 91–100 CrossRef CAS PubMed.
- A. S. Girgis, H. M. Hosni and F. F. Barsoum, Bioorg. Med. Chem., 2006, 14, 4466–4476 CrossRef CAS PubMed.
- R. Das, K. Bhattacharya, S. K. Samanta, B. C. Pal and C. Mandal, Cancer Lett., 2014, 351, 81–90 CrossRef CAS PubMed.
- S. Schott and A. Brüning, Gynecol. Oncol., 2014, 135, 342–348 CrossRef CAS PubMed.
- J. F. De Los Santos Jr, and J. M. Straughn, http://www.uptodate.com/contents/management-of-locally-advancedcervical-cancer.
- J. H. Maduro, E. Pras, P. H. Willemse and E. G. de Vries, Cancer Treat. Rev., 2003, 29, 471–488 CrossRef CAS.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- A. E.-F. G. Hammam, M. A. Sharaf and N. A. Abd El-Hafez, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2001, 40, 213–221 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copy of 1H NMR, 13C NMR, HRMS analysis, X-ray studies and anti-cancer bioassay data of the compound. See DOI: 10.1039/c4ra13433h |
| ‡ Deceased. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.