
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid, regioselective deuteration of dimethyl-2,2′-bipyridines via microwave-assistance†
Kitjanit Neranon and
Olof Ramström*
Department of Chemistry, KTH-Royal Institute of Technology, Teknikringen 30, S-10044 Stockholm, Sweden. E-mail: ramstrom@kth.se; Fax: +468 7912333
First published on 2nd December 2014
Abstract
Isotopically pure [D6]-dimethyl-2,2′-bipyridine derivatives were selectively and rapidly formed using microwave-assisted regioselective deuteration of the methyl moieties of the parent bipyridine in a deuterium oxide solution. For instance, [D6]-4,4′-dimethyl-2,2′-bipyridine was formed in quantitative yield within 15 minutes, in a simple and convenient process.
Introduction
Hydrogen–deuterium (H–D) exchange reactions at carbon have witnessed increasing attention since isotopically labeled compounds are recognized as having cumulative significance in NMR spectroscopy and mass spectrometry.1 For example, deuterated compounds are widely used as efficient tools for mechanistic investigations of chemical reactions, metabolic pathways, structural elucidations of biological macromolecules, and as internal standards for quantitative analyses of contaminants in foods and the environment.1,2 Therefore, efficient regioselective deuterium labeling approaches are of high importance for the preparation of deuterated reagents. For this reason, a range of deuteration methods has been reported, for instance based on transition metal-,3 enzyme-,7 acid-,4 or base-catalysis;5 super- or subcritical conditions;6 as well as microwave-enhanced exchange reactions.8 Many of these describe reactions performed at high temperature under pressure and/or for long periods of time, and the majority does not lead to chemo- and/or regioselective deuterations. In addition, these methods often require non-commercially available catalysts, strongly acidic or basic additives, and expensive D2 gas, and can furthermore lead to low deuteration efficiencies and regioselectivities. Consequently, the development of inexpensive, high-yielding, chemo- and/or regioselective deuteration processes under mild conditions is strongly desired. Herein, this objective has been addressed, where a simple and efficient method to deuterated bipyridine structures is reported.4,4′-Dimethyl-2,2′-bipyridine 1, 5,5′-dimethyl-2,2′-bipyridine 2, and 6,6′-dimethyl-2,2′-bipyridine 3 as shown in Fig. 1, are well-known and widely used bidentate ligands in applications of transition metal complexes, particularly in the inorganic photochemistry and supramolecular chemistry fields.9
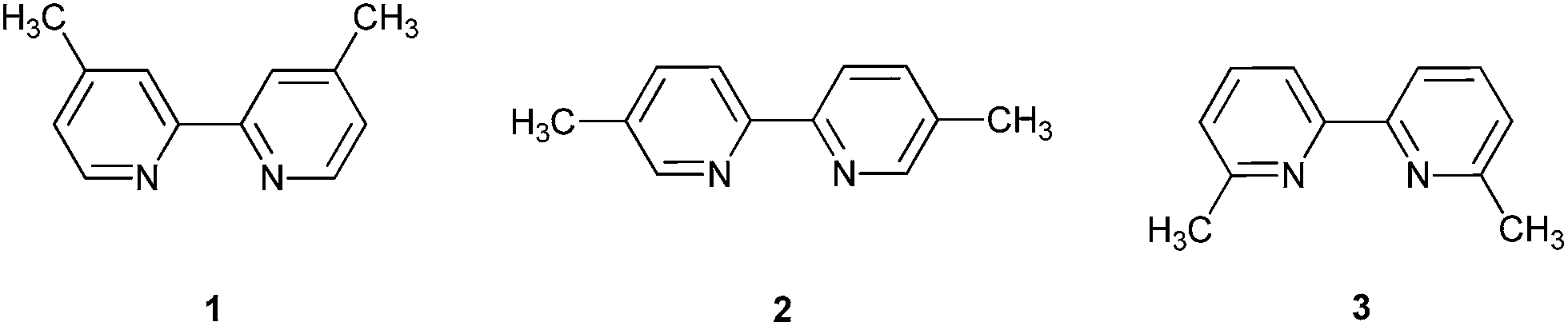 | ||
| Fig. 1 Bipyridine compounds examined. | ||
Examples of H–D exchange reactions of this class of compounds have also been reported. For example, Pavlik et al. reported that methyl groups at various positions of pyridine derivatives could be deuterated by a D2O–Na2CO3 solution at 110 °C from methylpyridine N-oxide derivatives.10 However, regioselective deuteration of the methyl groups without the introduction of deuterium on the pyridine ring could not be observed in this synthetic route. Browne and co-workers reported routes to regioselective deuteration of heteroaromatic compounds containing pyrazyl, pyridyl, 1,2,4-triazole, thienyl, methyl, and phenyl moieties based on subcritical aqueous media.6b The main deuterated product was in this case the fully deuterated [D12]-4,4′-dimethyl-2,2′-bipyridine 1-d12, whereas selective introduction of deuterium atoms on the 4- and 4′-methyl moieties could not be observed. However, although 1-d12 was obtained in good yield, the reactions required lengthy reaction times (4–6 days) as well as a high temperature (200 °C).
During the last decade, microwave-assisted organic synthesis has witnessed rapid growth, and has been widely used to accelerate different reactions.11 Enhanced selectivities have also been reported in some cases.11 For example, several examples of microwave-assisted deuterations in D2O have been reported,2d,8d of which some showed noticeable acceleration effects in conjunction to enhanced deuteration selectivities.12
The regioselective introduction of deuterium atoms into the 2,2′-bipyridine core, of special interest for spectroscopy applications, has however been limited. Current protocols generally involve time-consuming, multiple steps, can be of unfriendly nature, and may suffer from occasional, imperfect deuteration.6b,13 There is thus a lack of generally applicable, high-yielding, and low-cost deuteration procedures for this class of compounds. Furthermore, the motivation behind the interest in the deuteration of 2,2′-bipyridine ligands is their prospective applicability in the further investigation of supramolecular systems. Herein, we report a rapid, one-pot, regioselective deuteration method of 4,4′-dimethyl-2,2′-bipyridine (1), 5,5′-dimethyl-2,2′-bipyridine (2) and 6,6′-dimethyl-2,2′-bipyridine (3) in D2O under microwave irradiation to afford the deuterated products 1-d6, 2-dn (n=1–6) and 3-d6.
Results and discussion
Conventional heating conditions
The investigation of the direct H–D exchange reaction of bipyridine 1 was initiated by optimizing the reaction conditions using conventional heating. The H–D exchange reactions in D2O solutions were thus studied using various inorganic catalysts at different reaction temperatures for 24 h. The results from these exchange reactions are summarized in Table 1. Of the catalysts, Pd/C,14 and Na2CO3, known H–D exchange reaction catalysts for heterocyclic compounds,8d,10 and bipyridines,6b,13b did however not give any H–D exchange of compound 1 in D2O solutions (entries 1–4). In all cases, increasing the reaction temperature did not yield significant advantages over the reaction conducted at lower temperature (entries 1–4). Increasing the amount of 10% Pd/C from 10 wt% to 30 wt% had an insignificant effect on the deuterium incorporation, yielding a small amount of 1-d1 (10%, entry 5). Less than quantitative recoveries could be obtained in this case, due to difficulties in removing the substrate from the Pd/C catalyst. Increasing the amount of Na2CO3 from 10 wt% to >100 wt% failed to improve the deuterium incorporation with no desired 1-d6 product (entry 6). However, the use of 1 M NaOD/D2O provided slight H–D exchange at the methyl moieties of compound 1 (entry 7), while no H–D exchange was observed at the bipyridine ring.
|
||||
|---|---|---|---|---|
| Entry | Catalysta | Temperature (°C) | Recoveriesb (%) | % Dc |
| a 10% Pd/C (10 wt%) or Na2CO3 (10 wt%), and 1 M of NaOD/D2O (>100 wt%) were used.b Based on recovered yield.c Deuterium incorporation was determined by 1H NMR spectroscopy and ESI-MS.d 10% Pd/C (30 wt%) or Na2CO3 (>100 wt%) were used. | ||||
| 1 | Pd/C | Rt | Quant | — |
| 2 | Na2CO3 | 90 | Quant | — |
| 3 | Na2CO3 | Reflux | Quant | — |
| 4 | Pd/C | Reflux | Quant | — |
| 5d | Pd/C | Reflux | 87 | (10 1-d1) |
| 6d | Na2CO3 | Reflux | Quant | — |
| 7 | NaOD | Reflux | Quant | 9 |

Regioselectivities and yields/conversions of the deuterated products could be estimated using the unchanged bipyridine signal as internal reference in the 1H NMR spectrum, and the peak intensity of the initial substance 1 as internal reference in the ESI-MS spectrum, respectively. Although the H–D exchange reactions of compound 1 were unsuccessful under the reaction conditions listed in Table 1, these results suggested further investigation of the H–D exchange reaction in 1 M of NaOD/D2O as catalyst.
Microwave heating conditions
The H–D exchange reaction of bipyridine 1 in the presence of NaOD/D2O was next evaluated under microwave heating conditions (Table 2). The results were in this case very clear, and the exchange reaction proceeded more efficiently. Similar results as under conventional heating conditions were obtained until the temperature reached 150 °C, yielding low amounts of deuterated product, but increasing the temperature resulted in higher product yields. At 160 °C, up to 93% conversion could be recorded after 3 hours, but microwave irradiation at 170 °C for 15 min yielded excellent H–D exchange of all methyl protons (entry 7). In addition, no H–D exchange of the bipyridine ring protons could be observed under these conditions. Decreasing the amount of NaOD clearly showed reduced deuterium incorporation and 1-d6 product, accompanied by slightly decreased regioselectivity providing some 1-d4 and 1-d5. On the other hand, increasing the concentration of NaOD to 2 M caused explosion of the MW reaction vessel with the present setup and 1 M was therefore chosen as the optimal concentration.
|
|||||
|---|---|---|---|---|---|
| Entry | Time (min) | Temperature (°C) | Pressure (Bar) | Yieldb (%) | % Dc |
| a Reactions were performed in microwave reactor.b Based on recovered yield.c Deuterium incorporation was determined by 1H NMR spectroscopy and ESI-MS.d 0.1 M of NaOD/D2O was used.e 0.5 M of NaOD/D2O was used. | |||||
| 1 | 15 | 130 | 5 | Quant | — |
| 2 | 15 | 140 | 7 | Quant | 3 |
| 3 | 15 | 150 | 8 | Quant | 7 |
| 4 | 15 | 160 | 11 | Quant | 87 |
| 5 | 60 | 160 | 11 | Quant | 91 |
| 6 | 180 | 160 | 12 | Quant | 93 |
| 7 | 15 | 170 | 15 | Quant | 97 |
| 8d | 15 | 170 | 14 | Quant | 78 |
| 9e | 15 | 170 | 14 | Quant | 82 |

The 1H and 13C NMR spectra of deuterated product 1-d6 are displayed in Fig. 2 together with those of compound 1 for comparison. Upon H–D exchange, the singlet corresponding to the 4- and 4′-methyl moieties of compound 1 disappeared. Moreover, the two additional carbon satellite peaks of the methyl carbons in the 13C NMR spectrum revealed the characteristic C–D correlation pattern.
 | ||
| Fig. 2 (a) 1H NMR of compounds 1 and 1-d6 in CDCl3 solution. (b) 13C NMR of compounds 1 and 1-d6 in CDCl3 solution. | ||
To further confirm the identity of compound 1-d6 as deuterated, a crude reaction of compound 1 in methanol solution was analyzed by ESI-MS (Fig. 3a). The distribution is in this case very clear, with a significant signal of [1-d6 + Na]+ at 213.1267. Accordingly, compound 1-d6 was thus clearly obtained.
 | ||
| Fig. 3 ESI-MS spectra of H–D exchange reactions of (a) 1, (b) 2 and (c) 3 in methanol solution. | ||
In addition, the direct H–D exchange reaction of bipyridine 2 and 3 were evaluated under the optimal reaction conditions for both species using microwave irradiation at 190 °C for 3 hours with pressure 21 bar using ESI-MS in methanol solution. Their results are shown in Fig. 3b and c (1H and 13C NMR spectra see ESI†).
In the case of compound 3, as shown in Scheme 1, ESI-MS revealed a corresponding significant signal [3-d6 + Na]+ at 213.1283 along with a very small peak of compound 3. The 1H and 13C NMR spectra of compound 3-d6 demonstrated good H–D exchange of all methyl protons as quantitative yield with 93% deuterium incorporation (see ESI†).
 | ||
| Scheme 1 Optimal H–D exchange reaction of compound 3. | ||
In contrast to bipyridines 1 and 3, a distribution of deuterated products of bipyridine 2 was obtained with the deuterated compound 2-d4 as the major product (29%), together with lower amount of further deuterated products 2-d5 (23%), 2-d3 (19%), 2-d6 (14%), 2-d2 (10%), and 2-d1 (2%), respectively (Fig. 3b and ESI†). The results are shown in Scheme 2 as monitored by ESI-MS. Although several H–D exchange reaction conditions of compound 2 were considered by increasing the temperature and/or time, in which more deuterium incorporation was obtained in both cases, the fully deuterated products as in the case of bipyridine 1 or 3 could not observed. In analogy to bipyridines 1 and 3, however, deuteration took place at the methyl moieties of compound 2, and no H–D exchange of the bipyridine ring protons were found. One of the most important properties of alkylpyridines is the increased acidity of the hydrogens attached to the α-carbon of the side-chains, an effect strongest for the alkyl groups at position 2 and 4 of the pyridine ring, corresponding to positions 6 and 4 in the case of the 2,2′-bipyridine ring, respectively. The mobilities of the hydrogens in the α-positions of the side-chains were found to decrease as follows: 4-methylpyridine > 2-methylpyridine > 3-methylpyridine (pKa 30–37).15 Accordingly, the hydrogens of the 6,6′- and 4,4′-methyl substituents were more easily exchanged by deuterium than the hydrogens at the 5,5′-methyl positions.
 | ||
| Scheme 2 Optimal H–D exchange reaction of compound 2. | ||
Conclusions
In summary, a synthetic method for the regioselective deuteration of 4,4′-dimethyl-2,2′-bipyridine 1, 5,5′-dimethyl-2,2′-bipyridine 2, and 6,6′-dimethyl-2,2′-bipyridine 3 to (dn)-bipyridine under microwave-assisted direct H–D exchange is demonstrated. The process is straightforward, resulting in a high degree of isotopic purity, and untraceable amounts of side-products.Experimental section
General information
The deuterium contents of deuterium oxide was 99.9 atom %. 1 M NaOD/D2O solution was directly prepared by the dissolution of sodium metal (230 mg) in 99.9 atom % D2O (10 mL). 1H and 13C NMR spectra were recorded on a Bruker DMX 500 spectrometer at 500 MHz and 125 MHz. All NMR spectra were measured in CDCl3 (1H: δ = 7.26, 13C = 77.16) using the solvent residual signal as internal standard; chemical shifts (δ) are reported in parts per million (ppm) values and J values are given in Hertz (Hz). Microwave irradiation was performed with a Biotage® Initiator classic microwave reactor with a non-invasive sensor pressure control system; temperatures of the reaction mixtures were monitored by a perpendicularly focused IR temperature sensor and not by an internal temperature probe. High-resolution mass spectrometry was performed with an electrospray mass spectrometer (a Finnigan LCQ Advantage Max: LC-MS/MS).Acknowledgements
This work was supported by the Swedish Research Council, the Royal Institute of Technology, and the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/under REA grant agreement no 264645.Notes and references
- T. Junk and W. J. Catallo, Chem. Soc. Rev., 1997, 26, 401 RSC.
- (a) K. Liu, J. Williams, H.-R. Lee, M. M. Fitzgerald, G. M. Jensen, D. B. Goodin and A. E. McDermott, J. Am. Chem. Soc., 1998, 120, 10199 CrossRef CAS; (b) P. Mottier, I. Huré, E. Gremaud and P. A. Guy, J. Agric. Food Chem., 2006, 54, 2018 CrossRef CAS PubMed; (c) D. M. Marcus, M. J. Hayman, Y. M. Blau, D. R. Guenther, J. O. Ehresmann, P. W. Kletnieks and J. F. Haw, Angew. Chem., 2006, 118, 1967 CrossRef; (d) J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744 CrossRef CAS PubMed; (e) A. González-Antuña, P. Rodríguez-González, I. Lavandera, G. Centineo, V. Gotor and J. I. García Alonso, Anal. Bioanal. Chem., 2012, 402, 1879 CrossRef PubMed.
- (a) R. N. Garman, M. J. Hickey, L. P. Kingston, B. McAuley, J. R. Jones, W. J. S. Lockley, A. N. Mather and D. J. Wilkinson, J. Labelled Compd. Radiopharm., 2005, 48, 75 CrossRef CAS; (b) M. Yamamoto, K. Oshima and S. Matsubara, Heterocycles, 2006, 67, 353 CrossRef CAS; (c) C. W. Leung, W. Zheng, D. Wang, S. M. Ng, C. H. Yeung, Z. Zhou, Z. Lin and C. P. Lau, Organometallics, 2007, 26, 1924 CrossRef CAS; (d) K. Ishibashi and S. Matsubara, Chem. Lett., 2007, 36, 724 CrossRef CAS.
- (a) Y. Yang and R. F. Evilia, J. Supercrit. Fluids, 1999, 15, 165 CrossRef CAS; (b) K. Fodor-Csorba, G. Galli, S. Holly and E. Gács-Baitz, Tetrahedron Lett., 2002, 43, 3789 CrossRef CAS.
- (a) M. Okazaki, N. Uchino, N. Nozaki and K. Kubo, Bull. Chem. Soc. Jpn., 1995, 68, 1024 CrossRef CAS; (b) B. Lygo and L. D. Humphreys, Tetrahedron Lett., 2002, 43, 6677 CrossRef CAS; (c) J. Kalpala, K. Hartonen, M. Huhdanpää and M.-L. Riekkola, Green Chem., 2003, 5, 670 RSC.
- (a) M. Yamamoto, K. Oshima and S. Matsubura, Org. Lett., 2004, 6, 5015 CrossRef CAS PubMed; (b) W. R. Browne, C. M. O'Connor, J. S. Killeen, A. L. Guckian, M. Burke, P. James, M. Burke and J. G. Vos, Inorg. Chem., 2002, 41, 4245 CrossRef CAS PubMed.
- (a) N. G. Faleev, S. B. Ruvinov, M. B. Saporovskaya, V. M. Belikov, L. N. Zakomyrdina, I. S. Sakharova and Y. M. Torchinsky, Tetrahedron Lett., 1990, 31, 7051 CrossRef CAS; (b) A. Shulman, D. Sitry, H. Shulman and E. Keinan, Chem.–Eur. J., 2002, 8, 229 CrossRef CAS.
- (a) M. Yamamoto, K. Oshima and S. Matsubura, Chem. Lett., 2004, 33, 846 CrossRef CAS; (b) M. Takahashi, K. Oshima and S. Matsubara, Chem. Lett., 2005, 34, 192 CrossRef CAS; (c) K. Ishibashi, M. Takahashi, Y. Yokota, K. Oshima and S. Matsubara, Chem. Lett., 2005, 34, 664 CrossRef CAS; (d) A. Miyazawa, H. Shimodaira and Y. Kawanishi, Bull. Chem. Soc. Jpn., 2011, 84, 1368 CrossRef CAS.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. V. Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS; (b) V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Coord. Chem. Rev., 1994, 132, 1 CrossRef CAS.
- J. W. Pavlik, T. Vongnakorn and S. Tantayanon, J. Heterocycl. Chem., 2009, 46, 213 CrossRef CAS.
- C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71 CrossRef CAS PubMed.
- (a) A. Miyazawa, K. Saitou, K. Tanaka, T. M. Gädda, M. Tashiro, G. K. S. Prakash and G. A. Olah, Tetrahedron Lett., 2006, 47, 1437 CrossRef CAS PubMed; (b) A. Miyazawa, M. Tashiro, G. K. S. Prakash and G. A. Olah, Bull. Chem. Soc. Jpn., 2006, 79, 791 CrossRef CAS; (c) T. M. Gädda, X.-Y. Yu and A. Miyazawa, Tetrahedron, 2010, 66, 1249 CrossRef PubMed.
- (a) W. R. Browne and J. G. Vos, Coord. Chem. Rev., 2001, 219–221, 761 CrossRef CAS; (b) E. C. Glazer, B. Belyea and Y. Tor, Inorg. Chem. Commun., 2005, 8, 517 CrossRef CAS PubMed.
- T. Kurita, K. Hattori, S. Seki, T. Mizumoto, F. Aoki, Y. Yamada, K. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2008, 14, 664 CrossRef CAS PubMed.
- G. Jones, P. A. Claret and A. G. Osborne, Chemistry of heterocyclic compounds: quinolines part II, John Wiley & Sons Ltd., London, vol. 32, 1982 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR and ESI-MS spectra data. See DOI: 10.1039/c4ra13397h |
| This journal is © The Royal Society of Chemistry 2015 |