
Synthesis of dimethyl carbonate (DMC) based biodegradable nitrogen mustard ionic carbonate (NMIC) nanoparticles†
P. Thyriyalakshmi and
K. V. Radha*
Bio-Products Laboratory, Department of Chemical Engineering, A.C. Tech, Anna University, Chennai-25, Tamil Nadu, India. E-mail: radhavel@yahoo.com; Tel: +91-44-22359124
First published on 6th January 2015
Abstract
Chitosan-nitrogen mustard ionic carbonate nanoparticles (C-NMIC-Nps) were fabricated for the first time by an ionotropic gelation method. The preparation of C-NMIC-Nps involved four steps. In the first step, green reagent dimethyl carbonate (DMC) reacted with 2-dimethylethanolamine (DMEA) to form bis-(2-dimethyl amino-ethyl)carbonate (DAEC). In the second step, DAEC underwent cationization with (1-chloro-2,3 epoxy)propane to form NMIC with a stable carbonate backbone having a greater number of sorption sites. Later, NMIC was successfully cross linked with chitosan to form C-NMIC, which was later on used to form C-NMIC nanoparticles (C-NMIC-Nps) by ionotropic gelation. The chemical structure of NMIC and C-NMIC was identified by proton nuclear magnetic resonance (1H-NMR), 13C nuclear magnetic resonance (13C-NMR) spectroscopy and elemental analysis. The spherical shaped, smooth and uniform distribution of the C-NMIC-Nps was observed by scanning electron microscopy (SEM) and high resolution transmission electron microscopy (HRTEM). The average particle size distribution of the C-NMIC-Nps was found to be 14.4 ± 3.1 nm as observed using a particle size analyzer. The functional groups of DAEC, NMIC and C-NMIC were identified by Fourier transform infrared spectroscopy (FTIR). The conversion of the crystalline nature of chitosan to the amorphous state in C-NMIC-Nps was studied by X-ray diffraction (XRD). The thermal stability of the C-NMIC-Nps was confirmed by thermo gravimetric analysis (TGA). The good cell viability of C-NMIC-Nps was assessed by in vitro MTT assay using VERO cell lines. Overall, the results suggested that C-NMIC-Nps had been prepared successfully. The prepared C-NMIC-Nps will act as a nano carrier of growth factors for wound healing application.
1. Introduction
In modern polymer science a wide range of materials, such as natural or synthetic polymers, lipids, surfactants and dendrimers have been employed as drug carriers. Among these, polysaccharides have received increasing attention because of their outstanding physical and biological properties.1In recent years, naturally occurring carbohydrate based biopolymers chitin and chitosan have gained impetus because of their excellent biocompatibility.2 Since, chitin and chitosan exist with some unique properties like biodegradability and bioactivity, and have a variety of potential applications in biomedical and biotechnological fields. Chitosan is more efficient than chitin in terms of adsorption capacity due to the presence of large number of free amino groups on the chitosan chain.3
Chitosan is a poly cationic biopolymer obtained mainly by deacetylation of chitin, which is the second most abundant natural polymer after cellulose. It is a linear co-polymer of β-(1,4)-2-acetamido-2-deoxy-β-D-glucose and β-(1,4)-2-amine-2-deoxy-β-D-glucose.4 Chitosan has both reactive amino and hydroxyl groups, that can chemically alter its properties, under mild conditions. It is generally regarded as biocompatible, biodegradable and nontoxic biomaterial. Chitosan has been proved to have antifungal property. It is an attractive candidate for treating wounds by acting as a haemostat and initiates fibroblast proliferation at the wound site.5
Chitosan shows high crystallinity and high degree of hydrogen bonding leading to poor solubility in water and common organic solvents.6 In recent years, there has been a growing interest in the modification of chitosan and its utilization as diversified agent in pharmaceutical, waste water treatment, cosmetics, drug delivery, heavy metal chelation, heterogeneous catalysis and many other applications.7 Ionic or covalent crosslinking between the polymeric chains plays a vital role in the formation of nanoparticles.8
Cross linkers are molecules with two reactive functional groups that allow the formation of bridges between the polymeric chains.9 Cross linked chitosan is very stable and maintain their strength even in acidic and basic solutions. It leads to the change in crystalline nature of chitosan and enhance sorption properties.10
Chitosan is able to form nanoparticles which were harvested spontaneously under mild acidic conditions in contact with negatively charged sodium tripolyphosphate (TPP) ions by ionotropic gelation method. This type of ionic cross linking of chitosan is usually a typical non-covalent interaction, which is reversible and this may avoid the potential toxicity of the reagent.11
The chemical modification of chitosan affords a wide range of derivatives such as quaternary chitosan, carboxy alkyl chitosan, thiolated chitosan, sugar bearing chitosan, bile acid modified chitosan and cyclodextrin-linked chitosan. The derivatives of chitosan impart amphiphilicity, which is an important characteristic for the formation of self-assembled nanoparticles, potentially suited for biomedical applications.12
Among the several chitosan derivatives, chitosan with cationic moieties has been prepared by quaternization of the amino group or by grafting small molecules or polymer chains on to chitosan backbone. By introducing the cationic nature of chitosan several drug conjugate approaches have been developed towards therapeutic applications.13
Recently, chitosan acetate with both hemostatic and antimicrobial properties has been used for wound healing application.14 A good wound healing material should maintain a moist environment but allows the drainage of wound exudate and prevents infection at the wound site. Hydroxypropyl chitosan is used as a delivery vehicle for fibroblast growth factor that accelerates wound healing.15 Biocompatible and water soluble carboxymethyl chitosan nanoparticles has been studied for delivery of antibiotics.16
Nowadays sulfur and nitrogen (half-) mustards are extensively employed in inorganic synthesis, organic synthesis and in the preparation of numerous pharmaceutical intermediates. These compounds are toxic and the toxicity is strictly related to their highly reactive chlorine atom. The replacement of chlorine atom by a carbonate moiety with DMC17 an eco-friendly, biodegradable and nontoxic reagent resulted in sulfur and nitrogen half mustard carbonate analogues. These analogues showed to maintain the chemical behaviour of the parent chlorine compounds, while losing their toxic properties.18 Thus, the current study mainly focussed on the synthesis of nitrogen mustard ionic carbonate (NMIC).
The objective of the study is to develop a novel, facile route for the preparation of biodegradable chitosan-nitrogen mustard ionic carbonate nanoparticles (C-NMIC-Nps) with a stable carbonate backbone that offers a great promise as wound healing platform. In the present work, a novel DMC based NMIC was synthesized and cross linked with chitosan to produce C-NMIC. Herein, DMC provides a stable carbonate moiety to the newly synthesized NMIC. Then, a simple ionotropic gelation method was used to prepare the C-NMIC-Nps. The cationic and the hydrophilic property of NMIC, would enhanced the sorption properties of chitosan, making C-NMIC-Nps a promising material in therapeutics.
The synthesized NMIC, and the prepared C-NMIC and C-NMIC-Nps were characterized by various methods namely FTIR, 1H-NMR, 13C-NMR, elemental analysis, SEM, HR-TEM, particle size analysis, XRD and TGA. The biocompatibility of C-NMIC-Nps was assessed by in vitro cytotoxicity test (MTT assay).
2. Experimental methods
2.1 Materials
Analytical grade dimethyl carbonate (DMC), 2-dimethylethanolamine (DMEA), triethyl amine (TEA) and epichlorohydrin were purchased from Sigma Aldrich. Chitosan of low molecular weight (85% deacetylation) with viscosity >200 cps, sodium tripolyphosphate (TPP) with 85% purity and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] with 98% purity were also obtained from Sigma Aldrich. All other reagents were analytical grades and were used without any further purification. Milli-Q grade (resistivity 18.2 MΩ cm−1) water was used in all the experiments.2.2 Characterization of DAEC, NMIC, C-NMIC and C-NMIC-Nps
FTIR spectra of all the samples were recorded using Alpha Bruker spectrophotometer in attenuated total reflectance (ATR) mode in the range of 4000–400 cm−1. 1H-NMR and 13C-NMR spectra of the samples were recorded on a Bruker D8 advance 500 spectrometer at 25 °C using D2O and D2O/CF3COOD as solvents. Mass spectra were recorded on a (EI) JEOL JMS-AX 500. Elemental analysis was performed on a Heraeus CHN-O-Rapid analyser. XRD Patterns were done with a Bruker D8 Advance XRD machine using Cu-Kα (λ = 1.54 Å) source. The layer spacing of the samples was calculated according to the Bragg's equation. The diffraction angles 2θ were set between 2° and 75° incremented with a scanning angle of 1° min−1.Thermal stability was examined by the TGA of TA Perkin Elmer instrument at a heating rate of 20 K min−1 in the temperature range between 30–700 °C under nitrogen atmosphere. The average particle size distribution was determined using dynamic light scattering (DLS) analyzer with a Malvern Zetasizer Nano-S. For SEM analysis, the particles were sputter-coated with gold film using a sputter coater and imaged under SEM (TESCAN-VEGA3 SBU). For HRTEM studies of the nanoparticles, 5 μL of the sample was coated on a carbon-coated copper HRTEM grid, which was subsequently air dried and was analysed by a high resolution HRTEM (JEM 2100, Jeol, Peabody, MA, USA) operating at a high voltage of 200 keV. The selected area electron diffraction (SAED) pattern of the C-NMIC was also recorded using the same instrument.
2.3 Preparation of NMIC and C-NMIC
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str), 1496 (N–CH3 assy def), 1386 (CH2 def CH2–O), 1254 (ester C–O assy str), 1094 (ester C–O sym str), 864 (C–N str), 660 (CH2 rock). 1H-NMR (500 MHz, D2O): δH ppm 3.06 (12H, s, N–CH3), 3.38 (4H, t, J = 4.5, N–CH2), 3.91–3.95 (4H, m, O–CH2). 13C-NMR (500 MHz, D2O): δc 54 (N–CH3), 56 (N–CH2), 67 (O–CH2), 161 (CO). EI-MS (m/z): 207 (M + 2, 130%).O), 1386 (CH2 def CH2–O), 1336 (CO def of CHOH), 1162 (alcoholic C–O str), 1012 (CH2 def of CH2OH), 1254 (ester C–O assy str), 960 (OH def of CHOH), 834 (C–N str), 660 (CH2 rock), 602 (C–Cl str). 1H-NMR (500 MHz, D2O): δH ppm 3.2 (12H, s, N–CH3) 3.5–3.67 (8H, m, NCH2), 3.9–4.1 (4H, m, OCH2), 4.8–4.9 (2H, m, CH2–CH–CH2), 3.4–3.48 (4H, m, CH2Cl). 13C-NMR (500 MHz, D2O): δc ppm 54 (N–CH3), 55 (N–CH2–CH2) and (O–CH2), 63 (N–CH2–CH), 67 (CH2–CH–OH), 53 (C–Cl), 165 (CO). EI-MS (m/z): 391.2 (M+ − Cl, 100%).
O str), 1496 (N–CH3 assy def), 1386 (CH2 def CH2–O), 1254 (ester C–O assy str), 1094 (ester C–O sym str), 864 (C–N str), 660 (CH2 rock). 1H-NMR (500 MHz, D2O): δH ppm 3.06 (12H, s, N–CH3), 3.38 (4H, t, J = 4.5, N–CH2), 3.91–3.95 (4H, m, O–CH2). 13C-NMR (500 MHz, D2O): δc 54 (N–CH3), 56 (N–CH2), 67 (O–CH2), 161 (CO). EI-MS (m/z): 207 (M + 2, 130%).O), 1386 (CH2 def CH2–O), 1336 (CO def of CHOH), 1162 (alcoholic C–O str), 1012 (CH2 def of CH2OH), 1254 (ester C–O assy str), 960 (OH def of CHOH), 834 (C–N str), 660 (CH2 rock), 602 (C–Cl str). 1H-NMR (500 MHz, D2O): δH ppm 3.2 (12H, s, N–CH3) 3.5–3.67 (8H, m, NCH2), 3.9–4.1 (4H, m, OCH2), 4.8–4.9 (2H, m, CH2–CH–CH2), 3.4–3.48 (4H, m, CH2Cl). 13C-NMR (500 MHz, D2O): δc ppm 54 (N–CH3), 55 (N–CH2–CH2) and (O–CH2), 63 (N–CH2–CH), 67 (CH2–CH–OH), 53 (C–Cl), 165 (CO). EI-MS (m/z): 391.2 (M+ − Cl, 100%).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3OH/H2O, till the pH of the solution becomes neutral. The formed C-NMIC was dried in a vacuum oven at 55 °C overnight and stored in a desiccator. FTIR: νmax/cm−1 3280 (OH str and NH str of primary amine), 1642 (CO str of amide), 1564 (NH bends of amide), 1374 (sym angular deformation of CH3), 1318 (CO def of CH–OH), 1258 (ester assy C–O str), 1150, 1062, 1030 and 896 (sym str of C–O–C), 822 (C–N str), 798 (OH bend). 1H-NMR (500 MHz, D2O/CF3COOD): δH ppm 1.3 (br, 3H, s, NHCOCH3), 2.4–2.5 (br, 4H, d, (NH–CH2)), 2.9–3.2 (br, 4H, d, (N–CH2–CH(OH))), 3.3 (br, 12H, s, (N–CH3)), 3.5–4.9 (br, m, (2H, N–CH2–CH2–O), (H1, H2, H3, H4, H5, H6, H6′), (4H, OCH2), (1H, CHOH)). 13C-NMR (500 MHz, D2O/CF3COOD): δc ppm 22 (CH3CO), 52 (N–CH3), 54 (NH–CH2), 56 (N–CH2), 60 (C6), 61 (O–CH2), 63 (C2), 67 (CH2–CH–OH), 73 (C3), 74 (C5), 78 (C4), 105 (C1), 167 (CO).000 rpm for 30 min at 4 °C. The precipitated nanoparticles were once again re-suspended in water, sonicated, centrifuged and freeze dried.
:1 CH3OH/H2O, till the pH of the solution becomes neutral. The formed C-NMIC was dried in a vacuum oven at 55 °C overnight and stored in a desiccator. FTIR: νmax/cm−1 3280 (OH str and NH str of primary amine), 1642 (CO str of amide), 1564 (NH bends of amide), 1374 (sym angular deformation of CH3), 1318 (CO def of CH–OH), 1258 (ester assy C–O str), 1150, 1062, 1030 and 896 (sym str of C–O–C), 822 (C–N str), 798 (OH bend). 1H-NMR (500 MHz, D2O/CF3COOD): δH ppm 1.3 (br, 3H, s, NHCOCH3), 2.4–2.5 (br, 4H, d, (NH–CH2)), 2.9–3.2 (br, 4H, d, (N–CH2–CH(OH))), 3.3 (br, 12H, s, (N–CH3)), 3.5–4.9 (br, m, (2H, N–CH2–CH2–O), (H1, H2, H3, H4, H5, H6, H6′), (4H, OCH2), (1H, CHOH)). 13C-NMR (500 MHz, D2O/CF3COOD): δc ppm 22 (CH3CO), 52 (N–CH3), 54 (NH–CH2), 56 (N–CH2), 60 (C6), 61 (O–CH2), 63 (C2), 67 (CH2–CH–OH), 73 (C3), 74 (C5), 78 (C4), 105 (C1), 167 (CO).000 rpm for 30 min at 4 °C. The precipitated nanoparticles were once again re-suspended in water, sonicated, centrifuged and freeze dried.2.4 Cytotoxicity studies
 | (1) |
3. Results and discussion
3.1 Mechanism
DAEC was synthesized by the reaction of DMC with DMAE at 88 °C through BAC2 mechanism as given in Scheme 1. When DMEA attacks the carbonyl carbon of DMC, carboxy methylation reaction takes place resulting in the formation of the expected DAEC and by-product methanol.17 The second step in Scheme 1 represents the synthesis of NMIC by nucleophilic addition reaction. Herein, DAEC in the presence of acid catalyst attack the C3 carbon of epoxy ring of 1-chloro(2,3 epoxy)propane, resulting in the three member ring opening reaction. Covalent attachment of NMIC to chitosan through N-alkylation was shown in Scheme 2. Wherein, N-alkylation of chitosan with NMIC takes place through the liberation of HCl at pH 7–8.19 | ||
| Scheme 1 Formation of DAEC and NMIC. | ||
 | ||
| Scheme 2 Schematic structure newly synthesised C-NMIC. | ||
The successful attachment of NMIC to chitosan was confirmed by elemental analysis (Table 1). The presence of NMIC in the modified chitosan caused a decrease in the carbon and hydrogen content in the resulting C-NMIC material. However, the percentage of nitrogen increases with the addition of NMIC to the chitosan sequence of the modified polymer.20 From the results it can be inferred that the presence of NMIC as pendent chain in C-NMIC forms co-valent cross linking between its chloride atoms and amino groups present on the chitosan polymer backbone. In addition the decrease in the C/N ratio suggests that C-NMIC cross linking occurred successfully on the addition of NMIC. Thus chitosan was chemically modified by the successful incorporation of NMIC as specified in Scheme 2.
| Sample | Element (%) | |||
|---|---|---|---|---|
| C | H | N | C/N | |
| Chitosan | 40.43 | 6.57 | 7.17 | 5.63 |
| C-NMIC | 40.22 | 5.71 | 7.62 | 5.27 |
3.2 Characterization studies
O stretch of ester.21,33 The peaks due to asymmetric deformation of N–CH3 group and C–O asymmetric stretch of ester appeared at 1496 cm−1 and 1254 cm−1 respectively.22,26 The presence of alcoholic OH confirms the successful cationization of DAEC by the appearance of peak at 3464 cm−1 (Fig. S1(b)†).23 The peak at 602 was assigned to C–Cl stretching of the so formed CHPDAEC (NMIC).21,24
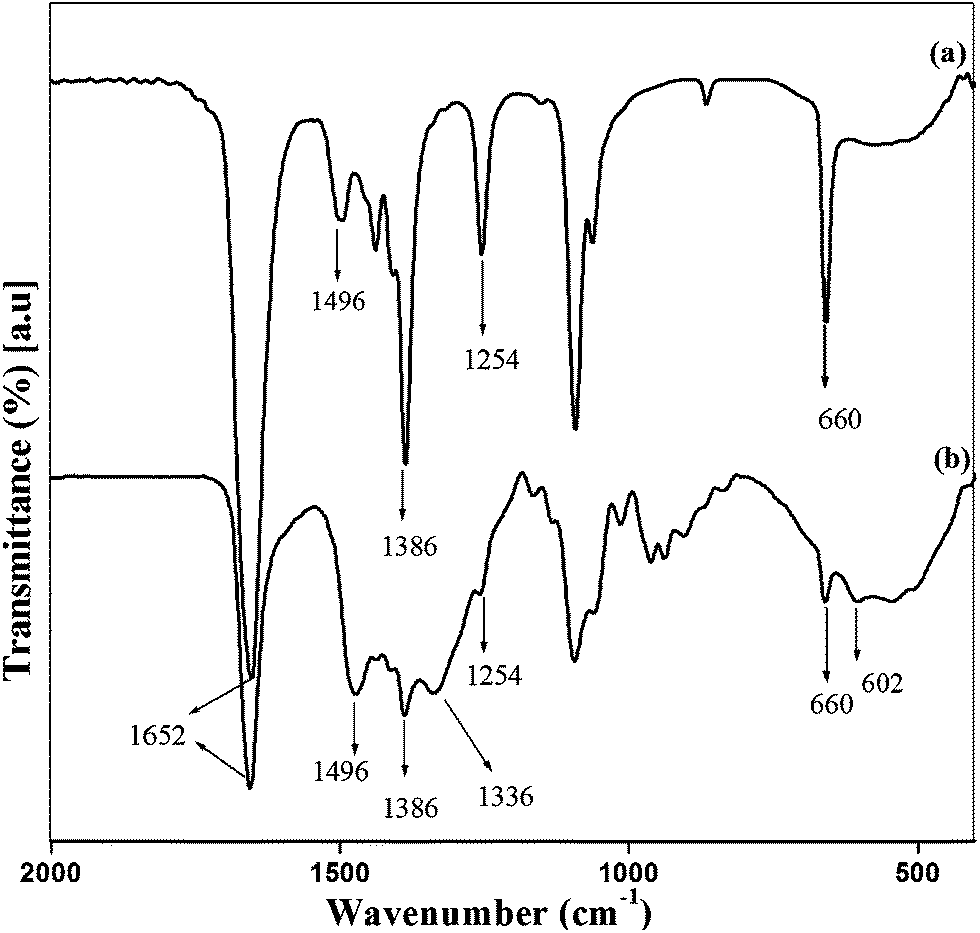 | ||
| Fig. 1 FTIR spectra of (a) DAEC and (b) NMIC. | ||
The unmodified chitosan spectrum (Fig. 2(a)) was similar to that of previously reported.25–28 In comparison to the supporting biomaterial chitosan, the FTIR spectra (Fig. 2(b)) of the prepared C-NMIC confirmed the successful addition of NMIC to chitosan through N-linkage by the appearance of new peak at 1546 cm−1.28 It should be noted that unlike chitosan, a characteristic band could be observed at approximately 1470 cm−1 in the C-NMIC sample, which was a clear evidence for the chemical modification of chitosan.25,29 Further, confirmation was done from the absence of peak at 602 cm−1, that arose from C–Cl stretching of NMIC.24
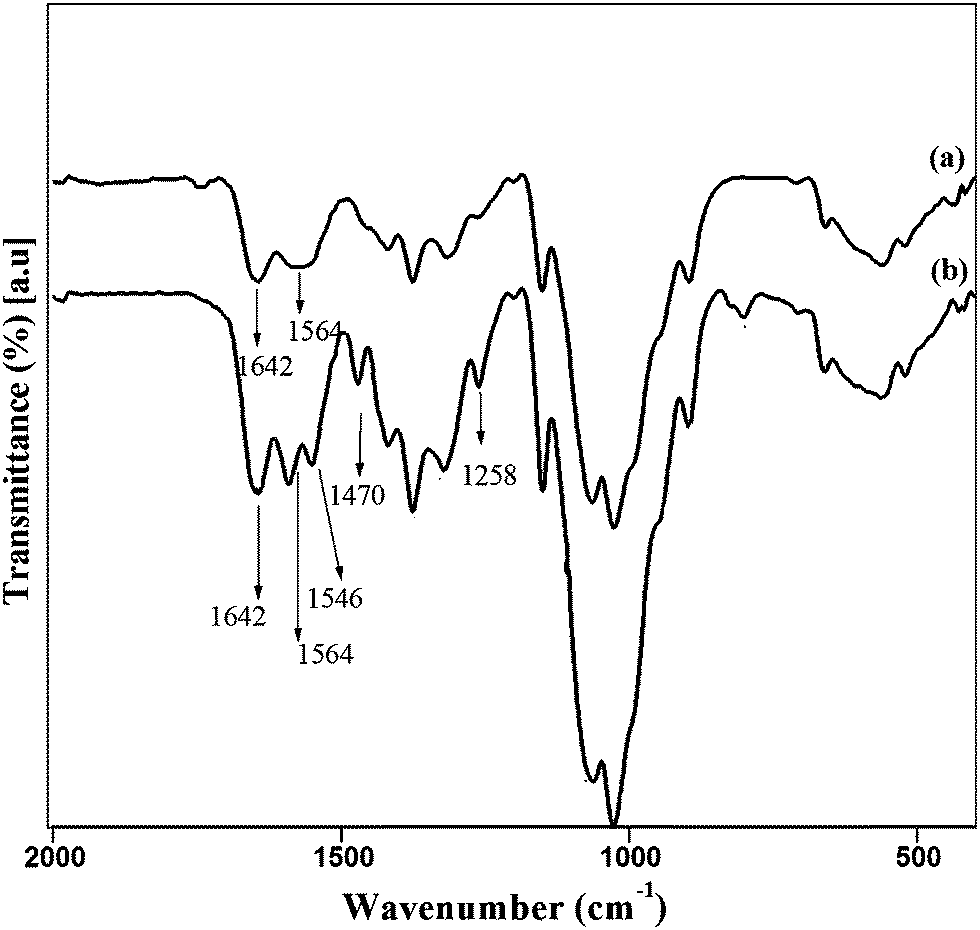 | ||
| Fig. 2 FTIR spectra of (a) chitosan and (b) C-NMIC. | ||
As shown in Fig. S3(c),† 1H-NMR of C-NMIC was similar to the one of chitosan except for the additional signals of N–CH3, and (NH–CH2) group protons at 3.3 ppm, 2.4–2.5 ppm respectively. These signals appeared as a singlet and doublet due to the presence of methyl and methylene protons.18,26 The doublet at 2.4–2.5 ppm showed the N-crosslinking of original chitosan with NMIC. Since a pH of 7–8 was maintained, N-alkylation takes place predominantly leading to 80–90% N-crosslinked C-NMIC.32
The 13C-NMR spectra (Fig. S4(a)†) indicated the formation of DAEC by the appearance of signals at δc 161, 67, 56 and 54 ppm for (CO), (O–CH2), (N–CH2) and (N–CH3) groups respectively.18,32–34 In addition to the signals found in DAEC, resonances at δc 67 and 53 ppm for the newly formed NMIC were attributed to the methine carbon of CH–OH32 and chloro carbon of C–Cl (Fig. S4(b)†).24
13C-NMR spectra of C-NMIC exhibited the chitosan signals, that was consistent with that as reported earlier.9,19,32 In addition signals at 167, 67, 61, 56, 54 and 52 ppm, were assigned to the NMIC carbonyl carbon, methine carbon of (CH–OH), methylene carbon of (O–CH2), (N–CH2), (NH–CH2) and methyl carbon of (N–CH3) respectively, which were an indication of neat formation of C-NIMC (Fig. S4(c)†).19,31,32
The EI mass spectra (Fig. S5(a) and (b)†) of the synthesized DAEC and NMIC showed molecular ion peak at (m/z) 207 and 391.2 respectively which was in good agreement with the calculated theoretical values.
4,39 that exhibits amorphous or disordered crystalline phase of chitosan. The decrease in chitosan crystallinity might be due to the weak ionic interaction of TPP which, diminishes the regularity of the extended C-NMIC crystal structure.20,35 But, the results implied that the crystalline domains of chitosan were not disrupted completely due to the presence of the active and flexible NMIC groups present in C-NMIC-Nps.7 The poly crystalline nature of the nanoparticles were further confirmed from the SAED pattern obtained from HRTEM analysis.From differential thermo gravimetric (DTG) curves (Fig. S8†), it was observed that the degradation temperature of chitosan in C-NMIC was lowered by 10 °C compared to original chitosan. This decreasing trend could be due to the random cross linking of NMIC having more number of polar groups with chitosan polymer backbone.43 C-NMIC-Nps (Fig. S8(c)†) showed two decomposition peaks at 260 °C and 370 °C respectively. This corresponds to the degradation temperature of chitosan and TPP.35 Once again herein, a further decrease in chitosan decomposition temperature would be due to the decrease in chitosan crystallinity that arises as a result of ionic cross linking of TPP with C-NMIC.19 On the basis of the results obtained from TGA and DTG, it can be stated that increase of polar groups and decrease in the crystalline domains caused the reduction in thermal stability.
 | ||
| Fig. 3 (a) Particle size distribution obtained from DLS (b) SEM image (c) HRTEM image, (d) lattice fringes in HRTEM and (e) SAED pattern of C-NMIC-Nps. | ||
3.3 Biocompatibility study
Biocompatible studies were done to evaluate the suitability of any polymer based nanoparticles used for biomedical applications. Biocompatibility of the C-NMIC-Nps was carried out by MTT assay using VERO cell lines. Cell viability of VERO cells with different concentrations of pure C-NMIC-Nps along with control (100%) was assessed by MTT assay. MTT was used to assess the cell viability as a function of redox potential. Actively respiring cells convert the water soluble yellow tetrazolium MTT to an insoluble purple formazan.It has been claimed that, the MTT formazan gives rise to extracellular deposits of needle-shaped crystals by exocytosis.38 These crystals were dissolved in DMSO. The resulting purple solution was spectrophotometrically measured. The effect of the nanoparticles on the cell proliferation was expressed as (%) cell viability.39–42
The results (Fig. S9†) exposed that the C-NMIC-Nps at 0.5 mg mL−1 dilution after 72 h showed remarkable biocompatibility to VERO cell lines with a maximum cell viability of 93.5%. A slight decrease in cell viability was observed as the concentration of the C-NMIC-Nps increased from (0.5–2.5 mg mL−1). This indicates the concentration dependent cytotoxicity of the nanoparticles.43 The significant increase in the cell viability of C-NMIC-Nps at all given dilutions with increasing time interval (12 h to 72 h) suggested a good development of cells within. Thus the positive time dependence property was a neat indication of the cell adherence and proliferation over the C-NMIC-Nps. The phase-contrast images of the viable cells in VERO cell lines seeded on control and C-NMIC-Nps after 72 h of incubation were shown in Fig. S10.† The results suggested that the C-NMIC-Nps has negligible cytotoxicity. Hence, the nanoparticles were biocompatible with the living cells and act as a potential material for wound healing application.
4. Conclusion
In conclusion, synthesis of novel NMIC, preparation of C-NMIC and C-NMIC-Nps and their characterization has been reported. Cross linking of NMIC with biodegradable carbonate moiety through N-alkylation with chitosan was verified using instrumental analysis. The morphology of the nanoparticles as observed by SEM and TEM images were found to be smooth and spherical in shape. The size of the C-NMIC-Nps was found to be in 10 nm range. The C-NMIC-Nps were proved to be non-toxic and showed high cell viability 93.5% at a concentration of 0.5 mg mL−1 highlighting their potential application in biomedical field as growth factor nano carrier for wound healing application.References
- R. Riva, H. Ragelle, A. Rieux, N. Duhem, C. Jerome and V. Preat, Adv. Polym. Sci., 2011, 244, 19–44 CrossRef CAS.
- L. Qian and H. Zhang, Green Chem., 2010, 12, 1207–1214 RSC.
- A.-H. Chen, S.-C. Liu, C.-Y. Chen and C.-Y. Chen, J. Hazard. Mater., 2008, 154, 184–191 CrossRef CAS PubMed.
- S. Sharma, P. Sanpui, A. Chattopadhyay and S. S. Ghosh, RSC Adv., 2012, 2, 5837–5843 RSC.
- W. G. Malette, H. J. Quigley, R. D. Gaines, N. D. Johnson and W. Gerald Rainer, Ann. Thorac. Surg., 1983, 36, 55–58 CrossRef CAS.
- K. G. Desai and H. J. Park, Drug delivery, 2006, 13, 375–381 CrossRef CAS PubMed.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- H. Jonassen and A.-L. Kjøniksen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 84, 1–4 CrossRef.
- M. Bodnar, J. F. Hartmann and J. Borbely, Biomacromolecules, 2005, 6, 2521–2527 CrossRef CAS PubMed.
- W. S. Wan Ngah, C. S. Endud and R. Mayanar, React. Funct. Polym., 2002, 50, 181–190 CrossRef.
- H. Liu and C. Gao, Polym. Adv. Technol., 2009, 20, 613–619 CrossRef CAS.
- J. H. Park, G. Saravanakumar, K. Kim and I. C. Kwon, Adv. Drug Delivery Rev., 2010, 62, 28–41 CrossRef CAS PubMed.
- S. K. Samal, M. Dash, S. V. Vlierberghe, D. L. Kaplan, E. Chiellini, C. V. Blitterswijk, L. Moroni and P. Dubruel, Chem. Soc. Rev., 2012, 41, 7147–7194 RSC.
- T. Dai, G. P. Tegos, M. Burkatovskaya, A. P. Castano and M. R. Hamblin, Antimicrob. Agents Chemother., 2009, 53, 393–400 CrossRef CAS PubMed.
- T. Dai, M. Tanaka, H. Ying-Ying and M. R. Hamblin, Expert Rev. Anti-Infect. Ther., 2011, 9(7), 857–879 CrossRef CAS PubMed.
- L. Zhao, B. Zhu, Y. Jia, W. Hou and C. Su, BioMed Res. Int., 2013, 2013, 1–7 Search PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- F. Aricò, M. Chiurato, J. Peltier and P. Tundo, Eur. J. Org. Chem., 2012, 17, 3223–3228 CrossRef.
- W. Sajomsang, S. Tantayanon, V. Tangpasthadol, M. Thatte and W. H. Daly, Int. J. Biol. Macromol., 2008, 43, 79–87 CrossRef CAS PubMed.
- E. C. N. Lopes, K. S. Sousa and C. Airoldi, Thermochim. Acta, 2009, 483, 21–28 CrossRef CAS PubMed.
- K. S. Babu, M. S. Kumari, L. K. Ravindhranath and J. Latha, Pharma Chem., 2013, 5(5), 123–130 CAS.
- M. M. J. Vijay Kumar, R. Yogananda, Snehalatha, H. Shameer, E. Jayachandran and G. M. Sreenivasa, J. Biomed. Sci. Res., 2009, 1(1), 1–10 CAS.
- R. Jayakumar, R. L. Reis and J. F. Mano, Drug Delivery, 2007, 14, 9–17 CrossRef CAS PubMed.
- D. Gor, P. Patel and P. S. Patel, J. Curr. Chem. Pharm. Sci., 2012, 2(4), 261–265 CAS.
- H. S. Mansur, A. A. P. Mansur, E. Curti and M. V. De Almeida, J. Mater. Chem. B, 2013, 1, 1696–1711 RSC.
- P. D. Chethan, B. Vishalakshi, L. Sathish, K. Ananda and B. Poojary, Int. J. Biol. Macromol., 2013, 59, 158–164 CrossRef CAS PubMed.
- P. Kithva, L. Grøndahl, D. Martin and M. Trau, J. Mater. Chem., 2010, 20, 381–389 RSC.
- C. L. Tien, M. Lacroix, P. Ispas-Szabo and M.-M. Mateescu, J. Controlled Release, 2003, 93, 1–13 CrossRef.
- W. Sajomsang, P. Gonil, S. Saesoo and C. Ovatlarnporn, Int. J. Biol. Macromol., 2012, 50, 263–269 CrossRef CAS PubMed.
- O. Kadkin, K. Osajda, P. Kaszynski and T. A. Barber, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1114–1123 CrossRef CAS.
- M. Sobczak, C. Dębek, E. Olędzka, G. Nałęcz-Jawecki, W. L. Kołodziejski and M. Rajkiewicz, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3904–3913 CrossRef CAS.
- W. Sajomsang, S. Tantayanon, V. Tangpasuthadol and W. H. Daly, Carbohydr. Res., 2009, 344, 2502–2511 CrossRef CAS PubMed.
- I. Kowalczyk, Molecules, 2008, 13, 379–390 CrossRef CAS PubMed.
- S. Daoudi, A. A. Othman, T. Benaissa and Z. O. Kada, Chem. Sci. Trans., 2014, 3(1), 281–291 Search PubMed.
- D. R. Bhumkar and V. B. Pokharkar, AAPS PharmSciTech, 2006, 7(2), 138–143 CrossRef PubMed.
- Q. Chen, A. Xu, Z. Li, J. Wang and S. Zhang, Green Chem., 2011, 13, 3446–3452 RSC.
- M. Li, Y. Wang, Q. Liu, Q. Li, Y. Cheng, Y. Zheng, T. Xi and S. Wei, J. Mater. Chem. B, 2013, 1, 475–484 RSC.
- Y. Liu, D. A. Peterson, H. Kimura and D. Schubert, J. Neurochem., 1997, 69, 581–593 CrossRef CAS.
- M. Liu, C. Wu, Y. Jiao, S. Xiong and C. Zhou, J. Mater. Chem. B, 2013, 1, 2078–2089 RSC.
- A. Cooper, N. Bhattarai, F. M. Kievit, M. Rossol and M. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 9969–9972 RSC.
- X. Zhong and F. Dehghani, Green Chem., 2012, 14, 2523–2533 RSC.
- G. Millotti, C. Samberger, E. Fröhlich, D. Sakloetsakun and A. Bernkop-Schnürch, J. Mater. Chem., 2010, 20, 2432–2440 RSC.
- N. S. Rejinold, M. Muthunarayanan, K. Muthuchelian, K. P. Chennazhi, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2011, 84, 407–416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra13290d |
| This journal is © The Royal Society of Chemistry 2015 |