
Exploring the substrate-assisted acetylation mechanism by UDP-linked sugar N-acetyltransferase from QM/MM calculations: the role of residue Asn84 and the effects of starting geometries†
Abstract
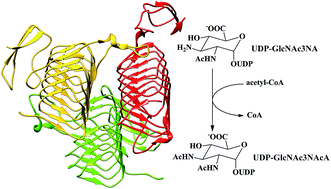
WlbB, one of the enzymes required for the biosynthesis of UDP-2,3-diacetamido-2,3-dideoxy-D-mannuronic acid (UDP-ManNAc3NAcA), is an N-acetyltransferase that catalyzes the N-acetylation of UDP-2-acetamido-3-amino-2,3-dideoxy-D-glucuronic acid (UDP-GlcNAc3NA) to form UDP-2,3-diacetamido-2,3-dideoxy-D-glucuronic acid (UDP-GlcNAc3NAcA). In this paper, based on the crystal structure, the detailed reaction mechanism of WlbB has been studied by using a combined QM/MM method. In particular, six snapshots taken from MD trajectories were used as the computational models to investigate how the starting geometries influence the calculation results. Our calculations suggest that the WlbB-catalyzed process involves two sequential steps. The nucleophilic attack of the C3-amino group of the substrate on the carbonyl carbon of acetyl-CoA occurs in concert with the departure of CoA from acetyl-CoA, generating a negatively charged CoA and a positively charged intermediate, which is inconsistent with the previous proposals that the catalytic reaction undergoes an oxyanion tetrahedral intermediate. Subsequently, the sulfur anion of CoA accepts the proton of the positively charged intermediate to yield the final product. Although Asn84 is not essential, it is important for promoting the catalysis by forming a hydrogen bond with the C3-amino group to position the lone pair of the electrons of the C3-amino group in an ideal orientation for nucleophilic attack and stabilize the transition states and intermediate. The cautious selection of initial geometries was found to be important for exploring the enzymatic mechanism and getting reliable energy barriers of the reaction pathways.
 Please wait while we load your content...
Please wait while we load your content...

