
Tuning the mechanical and morphological properties of self-assembled peptide hydrogels via control over the gelation mechanism through regulation of ionic strength and the rate of pH change†
Abstract
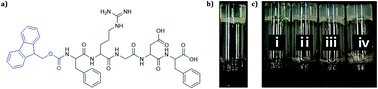
Hydrogels formed by the self-assembly of peptides are promising biomaterials. The bioactive and biocompatible molecule Fmoc-FRGDF has been shown to be an efficient hydrogelator via a π-β self-assembly mechanism. Herein, we show that the mechanical properties and morphology of Fmoc-FRGDF hydrogels can be effectively and easily manipulated by tuning both the final ionic strength and the rate of pH change. The increase of ionic strength, and consequent increase in rate of gelation and stiffness, does not interfere with the underlying π-β assembly of this Fmoc-protected peptide. However, by tuning the changing rate of the system's pH through the use of glucono-δ-lactone to form a hydrogel, as opposed to the previously reported HCl methodology, the morphology (nano- and microscale) of the scaffold can be manipulated.
 Please wait while we load your content...
Please wait while we load your content...

