
Immobilization of phosphomolybdic acid nanoparticles on imidazole functionalized Fe3O4@SiO2: a novel and reusable nanocatalyst for one-pot synthesis of Biginelli-type 3,4-dihydro-pyrimidine-2-(1H)-ones/thiones under solvent-free conditions†
Abstract
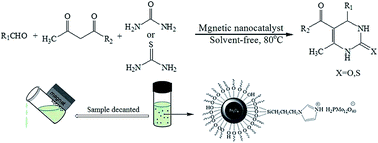
This article introduces Fe3O4@SiO2-imid-H3PMo12O40 nanoparticles as heterogenic catalyst for the synthesis of 3,4-dihydropyrimidinones under solvent-free condition. This reaction proceeds through acetoacetate esters and aldehyde derivatives followed by cyclisation with a urea to the dihydropyrimidinone. Excellent yields of dihydropyrimidinones are obtained within a short reaction time. The suggested method offers several advantages such as short reaction times, high yields, easy purification, a cleaner reaction and ease of recovery and reusability of the catalyst by a magnetic field. Also, the aforementioned nanocatalyst can be easily recovered by an external magnetic field and reused for subsequent reactions at least 5 times without noticeable deterioration in catalytic activity. Particle size distribution, surface area, magnetic properties and percent of leaching of H3PMo12O40 (PMA) after any reaction were investigated by dynamic light scattering (DLS), nitrogen physisorption method (BET) and Inductively Coupled Plasma (ICP) analyzer.
 Please wait while we load your content...
Please wait while we load your content...

